版权信息 书名:能源小分子的电化学转化
ISBN:978-7-115-69263-4
本书由人民邮电出版社发行数字版。版权所有,侵权必究。
您购买的人民邮电出版社电子书仅供您个人使用,未经授权,不得以任何方式复制和传播本书内容。
我们愿意相信读者具有这样的良知和觉悟,与我们共同保护知识产权。
如果购买者有侵权行为,我们可能对该用户实施包括但不限于关闭该帐号等维权措施,并可能追究法律责任。
版 权 著 王进 姚方磊 杨海朋 张成 李心驭 吴启康
责任编辑 贺瑞君
人民邮电出版社出版发行 北京市丰台区成寿寺路11号
邮编 100164 电子邮件 315@ptpress.com.cn
网址 http://www.ptpress.com.cn
读者服务热线: (010)81055410
反盗版热线: (010)81055315
内 容 提 要 本书系统阐述了碳、氢、氮等关键能源小分子的电化学转化路径,涵盖基本原理、研究进展与前沿应用3个方面。本书共分5章:第1章介绍电化学碳循环,主要包括CO2 还原反应与含碳小分子的电氧化;第2章介绍电化学氢循环,主要包括析氢反应、析氧反应、氢氧化反应及其可逆反应的核心原理,以及电解槽/燃料电池设计;第3章介绍电化学碳-氮-氢循环,主要包括电催化尿素合成与电催化氨基酸合成;第4章介绍先进催化剂,主要包括高熵合金、单原子催化剂的设计与应用;第5章总结本书内容,展望未来的发展方向,并介绍国家战略与愿景。
本书适合化学、化工、材料、能源及空间科学相关领域的科研人员、工程技术人员、高年级本科生及研究生阅读。
前 言 社会快速发展所带来的能源供给与环境污染问题,正推动着一场历史性的科技与工业变革。为解决该问题,必须从根本上改变高能耗、高排放的传统工业模式。同时,探测地外资源、建立月球基地、星际移民等长期规划的实现,依赖极端环境下原位利用地外资源合成燃料和生存物质,以实现物质与能量的循环自持。在此双重背景下,电化学转化技术脱颖而出。该技术具有环境友好、反应可控(通过电势调节反应速率和方向)、条件温和及成本效益高等优势,被视为连接可再生能源与绿色制造,乃至支撑地外生存的核心技术。
本书旨在系统阐述如何利用电化学转化这项强大技术,实现以碳(C)、氢(H)、氮(N)为核心的能源小分子的高效、定向转化。我们不仅关注该技术在地球层面的重大应用,如将二氧化碳(CO2 )转化为燃料与化学品、利用海水高效制取绿氢等,更将视野拓展至地外环境任务中的原位资源利用和生命保障系统。本书将深入探讨如何通过电化学碳-氮-氢循环,实现尿素、氨基酸等生命基础分子的合成,并分析这项技术在载人航天器、空间站生命支持系统,以及地外环境“就地资源利用”中的巨大潜力。此外,本书还将论述先进催化剂(如高熵合金、单原子催化剂)的合成与发展,为上述电化学反应提供催化材料基础。
本书围绕“元素循环”这个核心概念展开介绍,系统地整合基础原理、前沿应用与先进催化剂的知识框架,不仅能为化学、化工、材料、能源及空间科学相关领域的科研人员、工程技术人员、高年级本科生及研究生提供系统的基础理论,还能激发读者深入思考与探索电化学转化技术在未来能源变革与深空探索活动的应用。通往绿色地球与浩瀚星海的征程,或将从精准调控能源小分子转化的电化学反应中开启。
本书由王进教授负责整体构思、框架设计与统稿工作,并协调各章节内容,确保全书逻辑连贯。姚方磊、杨海朋负责撰写第3章、第5章;张成负责撰写第1章;李心驭负责撰写第4章;吴启康负责撰写第2章。在此,感谢本书各章作者投入的宝贵时间和付出的辛勤劳动,感谢作者所在课题组中的秦明鑫、叶媛媛、牛伟超、乐思思、李峰、陈孜恒等硕士研究生在文献整理、图表绘制等方面提供的帮助。
本书难免存在不足之处,敬请广大读者批评指正,我们将不胜感激。
著者
2025年12月
第1章 电化学碳循环:减碳导向的含碳小分子电化学转化 在能源结构深度调整的宏观背景下,构建以碳资源高效循环利用为核心的新型化工与能源体系,已成为实现可持续发展的关键路径之一。本章围绕“电化学碳循环”这一核心主题,系统介绍含碳小分子在电化学体系中的转化逻辑与技术框架,重点聚焦CO2 及相关含碳小分子的电催化还原(本书简称“电还原”)与电催化氧化(本书简称“电氧化”)过程,阐明其在减排增效、能源存储及绿色制造中的重要作用。通过对反应机理、产物生成路径(简称“产物路径”)、反应器设计及未来挑战的综合分析,为后续章节中不同元素循环的电化学转化研究奠定理论与方法基础。
1.1 电化学碳循环反应概述电化学碳循环反应是指在外加电能驱动下,通过电还原或电氧化过程实现碳元素在不同化学形态之间的可控转化,其核心在于将低价值、高排放量的含碳小分子转化为高附加值的燃料与化学品。这类反应不仅涉及多电子/多质子的复杂反应路径,还高度依赖催化剂表面结构、电极-电解质界面特性及反应微环境的协同调控。本节将从科学意义、基本反应原理及性能评估指标3个层面,对电化学碳循环反应进行系统概述,以明确其研究价值、理论基础与评价框架。
1.1.1 科学意义 在全球气候风险日益严峻、能源体系向低碳化加速转型的背景下,传统化工产业因具有高能耗、高排放的特点,被视为能源转型的关键难点领域,亟待建立新的绿色生产模式。电化学碳循环技术可利用电能驱动CO2 、醇或醛等含碳小分子的定向转化,为碳资源再利用和温室气体减排提供了一条兼具可持续性与战略价值的路径。
CO2 作为最主要的温室气体,全球年排放量已超360亿吨[1] 。通过CO2 还原反应(CO2 Reduction Reaction,CO2 RR),可将其转化为一氧化碳(CO)、甲烷(CH4 )、乙醇(C2 H5 OH)等燃料,以及甲酸(HCOOH)、乙烯(C2 H4 )等化学品,实现“减排”与“增值”的平衡。以C2 H4 为例,作为重要的基础化工原料,传统石脑油裂解路线生产每吨C2 H4 约排放3.2吨CO2 ,使化工行业的脱碳任务尤为艰巨。若通过CO2 RR实现C2 H4 的工业化生产,将改变C2 H4 生产过程中的碳流向,大幅削减CO2 的排放,为化工领域深度脱碳提供核心技术支撑[2] 。此外,CO2 RR等电化学转化技术可与光伏、风电等间歇性可再生能源高效耦合,将电能以化学能形式存储,缓解能源供需在时间与空间上的不匹配,对构建“电能-化学转化-燃料存储”的闭环体系具有不可替代的作用[3-4] 。
然而,电化学碳循环反应仍面临三大核心科学挑战:第一,C==O 的键能高达750kJ·mol-1 ,活化需克服较高能垒,导致反应速率较慢[5] ;第二,含碳小分子转化涉及多电子/多质子转移过程(如CO2 RR生成C2 H4 需12个电子转移),反应路径复杂,易形成多产物竞争(如CO、HCOOH、CH4 等),精准生成特定产物的难度(可用产物选择性指标衡量,详见第3章)非常高[6] ;第三,在水溶液体系中,阴极易发生竞争性析氢反应(Hydrogen Evolution Reaction,HER),导致目标产物的法拉第效率降低,进一步制约反应效率。
1.1.2 基本反应原理 电化学碳循环反应的本质在于电还原与电氧化两类过程的耦合,通过外加电势实现电子在电极与含碳小分子之间的定向转移,从而促使C—O、C—C、C—H等化学键发生断裂与重构,使低价值的碳源向高价值化合物转化,或将高排放路径转变为低排放路径。该反应并非单纯依赖电子供给即可完成,而是由电极-电解质界面环境、催化剂表面活性位点,以及反应物吸附与活化能垒的共同调节驱动。它能否发生,取决于热力学平衡电位,而反应速率与产物选择性则由电催化过程中的动力学特性控制。
从热力学本质来看,电化学碳循环反应的方向与限度由吉布斯自由能变(ΔG )决定:当ΔG <0时,反应可自发进行(如酸性条件下的CO氧化);当ΔG >0时,需通过外部电路施加电能(过电位)克服热力学能垒,驱动反应发生(如CO2 RR)。电子转移的速率与路径(动力学特性),则由催化剂表面的吸附-脱附平衡主导。以CO2 RR为例,CO2 分子首先需在催化剂活性位点(如Cu、Au等金属表面)发生物理吸附或化学吸附,通过注入电子打破稳定的线性O==C==O结构,生成带负电的活化中间体(如+ )结合,经历多步电子/质子转移(Proton-Coupled Electron Transfer,PCET),最终脱附生成目标产物(如CO、CH4 等)。这一过程中,催化剂对中间体的吸附强度至关重要:若吸附过强,会导致中间体难以进一步转化(如CO在Pt表面强吸附易造成催化剂中毒);若吸附过弱,则无法有效活化反应物(如CO2 在惰性金属表面难以生成稳定中间体),因此“适当的吸附强度”是实现高效转化的核心条件[7] 。
从界面反应特性来看,电化学碳循环反应依赖气-液-固三相界面的构建与调控:气相反应物(如CO2 、CO)需通过扩散进入电解液,在电极表面(固相催化剂)与电解质(液相)的界面处完成电荷转移与反应。界面微环境的参数(如pH值、离子强度、溶剂极性等)直接影响反应路径。此外,电极表面的电子传导速率、电解质中的离子迁移速率(如H+ 、OH- 、
综上,电化学碳循环反应的原理是热力学驱动(电位与吉布斯自由能)、动力学调控(催化剂与中间体)、界面特性(三相界面与微环境)三者的有机结合。电还原反应(如CO2 RR)通过电能输入实现碳的加氢还原与碳链增长,电催化氧化反应[如醇氧化反应(Alcohol Oxidation Reaction,AOR)]通过失去电子实现碳的“脱氢升级”与“官能团转化”,二者共同构成碳资源循环利用的电化学路径,为后续不同含碳小分子的定向转化提供了基础理论框架。
1.电还原反应(以CO2 RR为例) 在CO2 RR中,CO2 分子的惰性线性结构(O==C==O的键能高达750kJ·mol-1 )使其难以直接参与反应,因此催化剂表面的吸附活化过程是打破反应能垒的首要环节,而关键中间体*COOH(吸附态羧基)的生成与转化,更是决定反应路径、活性与产物选择性的核心。CO2 RR的本质是:在阴极催化剂表面,CO2 分子通过接受电子与质子,逐步转化为目标产物。CO2 RR的总反应及关键中间体路径因产物不同而异。以常见产物为例,典型反应方程式与相对于可逆氢电极(Reversible Hydrogen Electrode,RHE)的理论平衡电位如下[6,8] 。
(1)生成CO:CO2 +2H+ +2e- →CO+H2 O(E 0 =-0.106V)。
(2)生成HCOOH:CO2 +2H+ +2e- →HCOOH(E 0 =-0.144V)。
(3)生成CH4 :CO2 +8H+ +8e- →CH4 +2H2 O(E 0 =0.169V)。
(4)生成C2 H4 :2CO2 +12H+ +12e- →C2 H4 +4H2 O(E 0 =0.084V)。
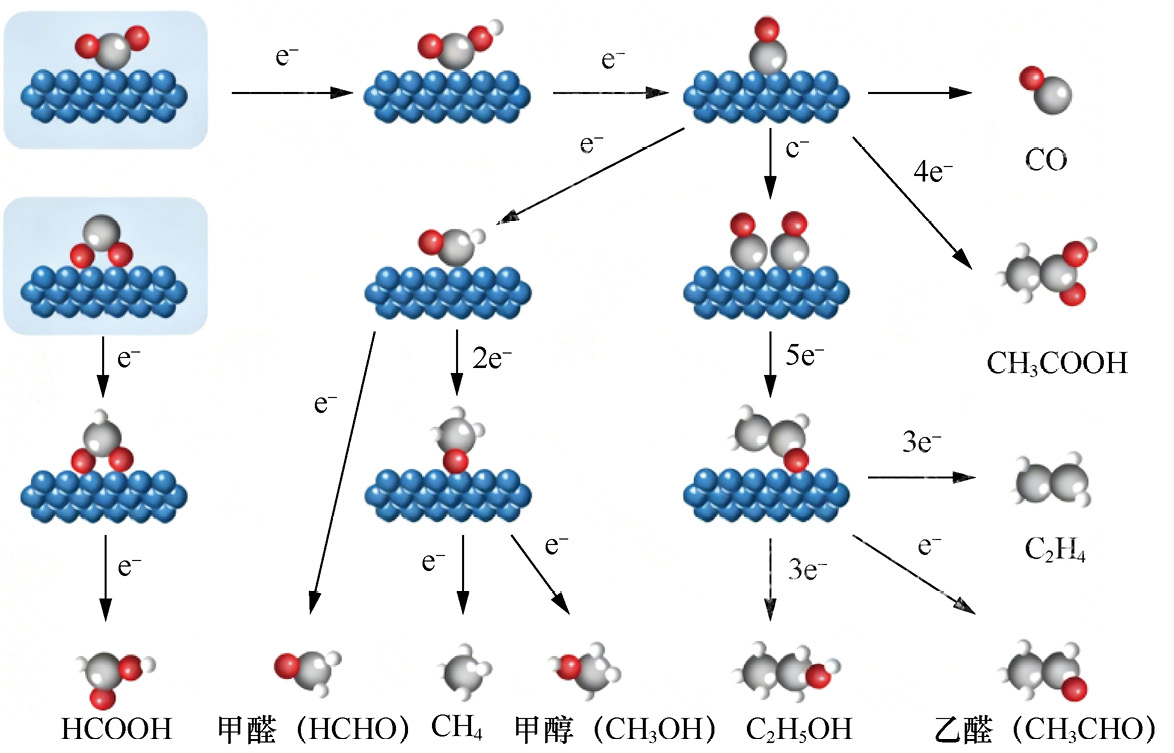
反应过程中,CO2 分子首先在催化剂表面吸附活化,生成*COOH,这通常是CO2 RR的决速步(Rate Determining Step,RDS)。随后,*COOH通过两种路径分化:若*COOH接受电子质子,转化为*CO,进一步还原可生成CO或C2+ 产物(如*CO+*CO→*OC—CO→C2 H4 );若*COOH直接质子化,则生成*HCOO,最终转化为HCOOH(见图1-1)[9] 。此外,关键中间体的吸附强度将直接决定反应路径。若催化剂对*COOH吸附过强,*COOH难以发生后续转化,易在催化剂表面堆积,不仅堵塞活性位点,还可能发生过度还原,生成积碳,导致催化剂失活;即使*COOH转化为CO,过强的CO吸附也会阻碍其脱附或二聚。例如,在Pt基催化剂表面,*CO的吸附能约为-1.8eV,几乎无法脱附,最终导致催化剂“CO 中毒”,CO2 RR 活性急剧下降[10] 。若催化剂对*COOH的吸附过弱(如部分惰性金属催化剂),*COOH生成后易从催化剂表面脱附,无法参与后续反应,导致CO2 RR活性极低。同时,吸附过弱意味着CO2 分子活化不充分,大量未活化的CO2 会直接从反应体系中逸出,反应物利用率显著降低[11] 。因此,通过调控(如合金化、掺杂、缺陷工程)催化剂电子结构,可以优化*COOH的吸附强度,实现关键中间体的适度吸附。例如,在Cu基催化剂中引入Zn(生成Cu-Zn合金),可优化*COOH的吸附能,既保证*COOH高效转化为CO,又避免CO过度吸附,实现合成气的可控合成[12] ;而在CuInS2 纳米片上构建的丰富S空位有效调节了金属活性位点周围的局部电子密度,诱导*COOH从竞争性吸附转变成特异性吸附,显著提升*HCOO的生成速率[13] 。
图1-1 CO2 RR的关键中间体路径示意图[9]
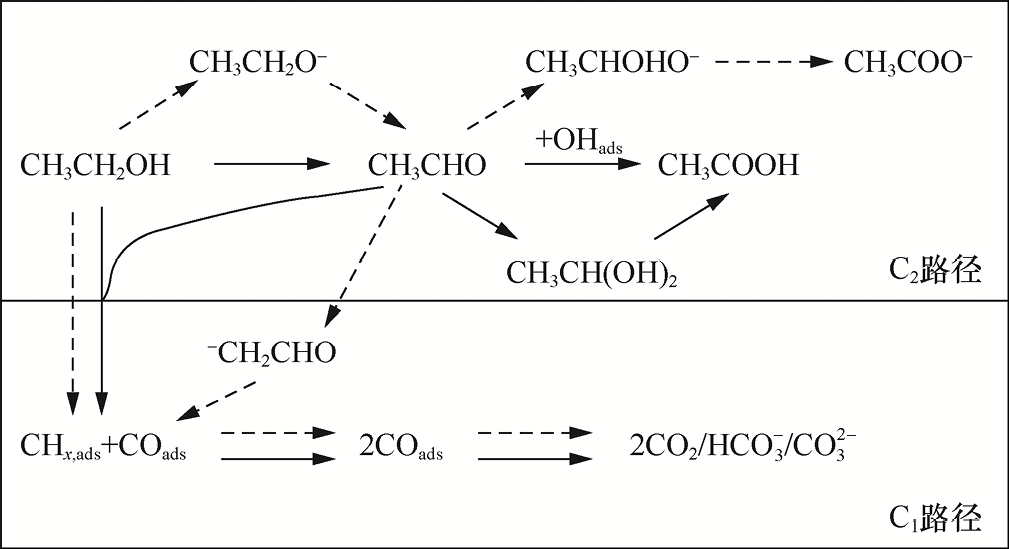
2.电氧化反应(以AOR为例) 含碳小分子的电氧化反应是通过在电极表面失去电子,实现碳中心氧化态升高与碳链转化/升级的核心过程,最终生成的是醛、羧酸、酮等高附加值含氧化合物,甚至可进一步断裂C—C,生成C1 产物(如CO2 、HCOOH)。该反应在燃料电池、精细化工合成等领域具有重要应用价值。其中,乙醇氧化反应(Ethanol Oxidation Reaction,EOR)作为多碳AOR的典型代表,因涉及C1 与C2 两条竞争反应路径(见图1-2),且产物选择性可通过催化剂与反应条件调控,成为研究电氧化反应机理的核心模型体系[14-15] 。从反应热力学与电子转移特性来看,EOR的总反应与关键产物的理论平衡电位(相对于RHE)清晰地反映了不同氧化路径的能量需求,其不同路径对应的反应方程式与理论平衡电位如下。
(1)C1 路径(碳链断裂)。C2 H5 OH在氧化过程中发生C—C断裂,生成单碳产物,核心反应包括生成HCOOH和CO2 。
生成HCOOH(C1 部分氧化):C2 H5 OH+3H2 O→2HCOOH+8H+ +8e- (E 0 ≈0.03V,相对于RHE)。该反应涉及8个电子转移,双碳骨架完全断裂为两个HCOOH分子,C氧化态升至+2价。
生成CO2 (C1 完全氧化):C2 H5 OH+3H2 O→2CO2 +12H+ +12e- (E ⁰ ≈ 0.06V,相对于RHE)。该反应涉及12个电子转移,是EOR的终极氧化路径,C氧化态达到+4价,需克服C—C断裂能垒(约347kJ·mol-1 ),能量需求相对更大。
(2)C2 路径(碳链保留)。C2 H5 OH分子保留双碳骨架,逐步脱氢氧化,生成含氧化合物,核心反应包括生成CH3 CHO和CH3 COOH。
生成CH3 CHO(部分氧化):C2 H5 OH→CH3 CHO+2H+ +2e- (E 0 =0.098V,相对于RHE)。该反应仅涉及2个电子转移,氧化态从C2 H5 OH中的C(-2价,平均)升至CH3 CHO中的C(-1价,平均),是能量需求较小的浅度氧化过程。
生成CH3 COOH(完全氧化,C2 终点):C2 H5 OH+H2 O→CH3 COOH+4H+ +4e- (E 0 =0.082V,相对于RHE)。该反应涉及4个电子转移,氧化态进一步升至CH3 COOH中的C(0价,平均),需引入水分子,令其参与氧原子补充,属于深度氧化但保留碳链的路径。
EOR可被视为由连续脱氢、关键中间体演化及碳链结构调控这3部分协同推进的复杂过程,不同反应路径均源自关键中间体的生成方式及其后续转化行为。整个机理通常可划分为以下两个主要阶段,而催化剂表面活性位点及界面环境是决定路径分化与产物选择性的关键因素。
图1-2 EOR中的反应机理。实线箭头表示低pH值下的机理,虚线箭头表示高pH值下的机理[15]
(1)C2 H5 OH吸附与初始脱氢(通用起始步骤)。反应起始于C2 H5 OH分子在催化剂表面发生吸附,可通过—OH或—C2 H5 基团与金属位点形成化学键。对于以Pt、Pd为代表的贵金属体系,C2 H5 OH的羟基氧通常与金属零价位点(如Pt0 、Pd0 )发生配位,从而实现稳定吸附。随后,C2 H5 OH会失去α-H,生成吸附态中间体*CH3 CHOH,这一步常被视为EOR的主要动力学瓶颈之一,且该中间体的能垒大小直接决定整体反应速率。以Pt催化剂为例,α-H的断裂能垒通常为0.3~0.5eV,而通过引入Ru、Sn等共催化元素可将该能垒降低至0.2~0.3eV,从而显著加速初始脱氢步骤[16] 。
(2)中间体转化与路径分化(C1 /C2 路径关键分支点)。*CH3 CHOH进一步脱氢后生成吸附态*CH3 CO(乙酰基),该物种(Species)被普遍认为是EOR中决定反应走向的关键公共中间体,也是C1 路径与C2 路径发生分化的核心分支点[17] 。在此基础上,反应将根据催化剂对*CH3 CO的吸附强度以及界面氧化性与亲核物种供给情况,分别沿C1 路径或C2 路径继续演化。
C1 路径启动:若催化剂对*CH3 CO的吸附强度过大,或界面存在高氧化态金属位点(如Pt4+ 、Ni3+ ),*CH3 CO会进一步发生C—C断裂。一种机制是*CH3 CO在高氧化态位点作用下,生成*CO(羰基)与*CH3 (甲基),*CH3 继续脱氢氧化,生成*CO,最终*CO与*OH反应生成CO2 (C1 完全氧化);另一种机制是*CH3 CO水解生成*HCOO(甲酸根),脱附后生成HCOOH(C1 部分氧化)。
C2 路径维持:若催化剂对*CH3 CO的吸附强度适中,且界面存在充足的OH- (碱性电解质)或H2 O,*CH3 CO会与吸附态*OH发生亲核反应,生成*CH3 COO(乙酰氧基),进一步质子化后脱附为乙酸(CH3 COOH);若催化剂对*CH3 CO的吸附较弱,*CH3 CO会直接失去一个H+ 和e- ,以CH3 CHO形式脱附,完成C2 部分氧化。
1.1.3 性能评估指标 在电化学碳循环反应的研究与应用中,反应效率、催化活性、能耗与长期稳定性共同决定技术的可行性与工业化潜力。为避免评价标准差异导致的研究结论偏差,需建立一套统一、科学的核心评估指标体系,从电子利用效率、反应速率、能耗成本及系统耐久性4个维度,全面量化催化体系与反应过程的综合性能。本小节对法拉第效率(Faraday Efficiency,用FE表示)、电流密度(Current Density,用j 表示)、过电位(Over Potential,用η 表示)、稳定性(Stability)这4个指标展开详细阐述,为本章的性能对比与技术分析提供统一基准。
1.法拉第效率 法拉第效率是电子利用效率与产物选择性的核心标尺,本质是目标产物电子消耗占比,核心价值在于量化电子在电催化反应中的定向利用程度。在电化学碳循环反应中,电子作为核心反应物,若大量流向副反应(如HER),不仅会降低目标产物的产率,还会造成能源浪费。因此,法拉第效率是评估催化剂的产物选择性与反应路径可控性的首要指标。
(1)定义与计算。法拉第效率的计算公式基于法拉第定律(电解过程中电极上沉积的物质量与通过的电量成正比)确定,需明确各参数的物理意义与测定方法:
其中,n 表示电子转移数,v 表示产物生成速率,I 表示总电流,F 表示法拉第常数。
电子转移数n 需根据目标产物的具体反应方程式精准确定。例如,CO2 RR生成CO时,CO2 →CO需要2个电子(n =2);生成C2 H4 时,2CO2 →C2 H4 需要12个电子(n =12)。若产物为混合体系(如CO与C2 H4 共存),需分别计算各产物的n 值并叠加电子消耗总量。
产物生成速率v 需通过精准检测手段测定。气态产物(如CO、C2 H4 )常用气相色谱(Gas Chromatogram,GC,如Agilent 7890A)结合气体流量控制器计算生成速率;液态产物(如HCOOH、CH3 OH)常用高效液相色谱法(High Performance Liquid Chromatography,HPLC,如Waters e2695)或核磁共振(Nuclear Magnetic Resonance,NMR)定量分析;微量产物可采用原位红外光谱或微分电化学质谱实时监测。
总电流I 需通过电化学工作站(如CHI 760E、Autolab PGSTAT302N)精准采集,且需扣除背景电流(如电解质自身分解电流),避免非反应电流导致计算得到的法拉第效率偏高。
(2)行业标准与应用场景。在电化学碳循环领域,法拉第效率可作为评估催化剂性能和反应体系可控性的关键指标。根据研究与应用的不同阶段,可对法拉第效率的期望水平作经验性区分:在基础研究阶段,更关注催化剂的产物选择性和反应路径的可控性,因此要求法拉第效率较高以证明电子定向利用效果;在中试或放大阶段,需要兼顾副产物产生与分离成本,因此期望法拉第效率在实验条件下持续保持高水平;在工业化或大规模生产条件下,高法拉第效率是实现能源高效利用和降低系统运行成本的重要因素。
在具体催化体系中,不同产物的法拉第效率通常存在差异。例如,对于单碳产物(如CO)体系,电子利用效率往往较高,表现出较好的产物选择性;而对于多碳产物(如C2+ 产物)体系,法拉第效率受反应路径复杂性及副反应影响,可能相对较低,需要通过催化剂设计和反应条件优化进一步提升。此外,在水系电解环境中,HER通常与CO2 RR竞争,尤其在酸性或中性电解质中,二者的启动电位可能部分重叠。这意味着测定法拉第效率时需注意扣除背景电流,并采用对照实验或同位素标记等手段提高测定可靠性,从而更准确地反映目标产物的电子利用效率。
2.电流密度 电流密度是衡量电催化体系活性及其放大潜力的关键指标,表征在单位电极面积与单位时间内的电子转移能力,直接反映反应速率与可达产量。较高的电流密度意味着在相同面积条件下可获得更高的产物生成速率,是推动电化学碳循环技术由实验室研究迈向工业化应用的重要基础。
(1)定义与分类:从“表观活性”到“本征活性”。电流密度需根据评估目的分为两类,以消除电极结构差异对活性判断的干扰。
几何电流密度(Geometric j ):基于电极的表观面积(如1cm2 的圆形电极)计算,单位为mA·cm-2 或A·cm-2 ,是评估反应器整体产能的直观指标。
电化学活性面积(Electrochemical Active Surface Area,ECSA)归一化电流密度(ECSA-Normalized j ):基于电极的电化学活性面积[1] -2 (ECSA),用于评估催化剂的本征活性(单位活性位点的催化能力)[18] 。
例如,两种Cu基催化剂的几何电流密度均为200mA·cm-2 ,但ECSA分别为10cm2 和20cm2 ,则前者的本征活性[20mA·cm-2 (ECSA)]是后者[10mA·cm-2 (ECSA)]的2倍,说明其活性位点的催化效率更高。
(2)测试条件与工业化要求。在电化学碳循环技术中,电流密度是评估催化剂活性和反应性能的重要指标。为了保证数据的可靠性与可重复性,在测定电流密度的同时需要控制关键反应参数,如反应温度、溶液浓度、反应物流速等。这些控制有助于减少实验误差,使不同实验之间的数据具有可比性。
从工业应用角度来看,不同产物对电流密度的需求存在差异。生成单碳产物(如CO、HCOOH)时,反应路径相对简单,适中的电流密度即可满足产能要求;生成多碳产物(如C2 H4 、C2 H5 OH)时,由于涉及多电子转移与C—C偶联,通常需要更大的电流密度以提高产量和效率;而某些电氧化反应(如生成酸类产物的AOR)由于反应动力学较快,工业应用条件下可承受较大电流密度。总体而言,工业化导向下的电流密度设定应结合产物类型、反应动力学特征及整体系统效率进行合理规划,而非依赖单一固定数值。
3.过电位 过电位是能耗与反应能效的核心指标,是衡量电催化反应能耗的关键参数。由于实际反应中存在活化能垒、电阻损耗等,需施加高于理论平衡电位的电压才能驱动反应发生,过电位越低,反应所需的电能越少,能效越高,直接决定技术的运行成本。
(1)定义与计算逻辑:从“热力学电位”到“实际电位”。过电位的计算公式需明确理论平衡电位(E equilibrium )与实际反应电位(E actual )的定义与获取方法:
注意,绝对值表示过电位为正值,仅反映能量消耗的大小,与反应方向无关。
理论平衡电位是基于热力学计算的反应电位,通常以RHE为参比电极,需根据反应方程式与能斯特方程(Nernst Equation)校正pH值。例如,CO2 RR生成CO的理论平衡电位为-0.106V(相对于RHE,pH=7),而在pH=1的酸性条件下,根据能斯特方程(E =E 0 +0.059pH),理论电位会偏移至-0.46V(相对于RHE)。
实际反应电位是指通过线性扫描伏安法(Linear Sweep Voltammetry,LSV)或塔费尔曲线(Tafel Plot)测定的起始反应电位或特定电流密度下的电位。例如,评估CO2 RR催化剂的过电位时,通常取电流密度为10mA·cm-2 时的实际反应电位与理论平衡电位的差值,该电流密度下的过电位更能反映催化剂的活化能力。
(2)分类与能效关联。过电位又可分为活化过电位(η activation )与欧姆过电位(η ohmic )。
活化过电位源自反应本身的活化能垒,是总过电位的主要构成部分之一。通常,活化过电位通过塔费尔分析进行评估:在其他条件相同的情况下,塔费尔斜率(Tafel Slope)越小,说明反应所需克服的活化能垒越低,电催化过程更容易进行。在CO2 RR领域,不同催化剂体系的反应路径复杂度不同,因此其塔费尔斜率往往具有可区分的特征。例如,某些贵金属介导的单碳产物路径相对简单,通常表现出较小的塔费尔斜率,而具有多步电子转移与C—C偶联的体系往往呈现更大的塔费尔斜率,需要更大的驱动力才能维持相同反应速率。
欧姆过电位主要由电解质溶液的离子电导率、反应器结构、电极连接电阻等因素共同决定。该过电位可以通过电化学阻抗谱(Electrochemical Impedance Spectroscopy,EIS)得到,并在实际测试中通过欧姆降(Ohmmic Drop,又称IR降)补偿进行修正。
(3)能效换算关系。过电位会直接影响系统的能耗,因为电解过程所需能量与施加的电压成正比。当反应所需的过电位降低时,在相同电流密度下系统的总体能耗会明显下降,有利于提升能效、减少运行成本。在电化学转化的实际应用场景中,通常希望在中等电流密度下(如数十毫安每平方厘米量级)实现尽可能低的过电位,以改善整体能量利用效率。对于拟用于规模化应用的催化体系,常见的目标是将过电位控制在适合长期运行且具有经济性的范围内,从而兼顾反应动力学、产物选择性和能效。
4.稳定性 稳定性是长期运行与系统可靠性的关键保障,也是评估电化学碳循环技术耐久性的核心指标,直接决定了反应器的维护成本与使用寿命。稳定性包括电化学稳定性(反应活性与产物选择性的长期保持)与结构稳定性(形貌、成分、晶相的不变性)。
(1)电化学稳定性:从“短期测试”到“长期考核”。电化学稳定性通常采用恒电位或恒电流的计时测试进行评估,通过观察电流或电位随时间的变化判断催化体系是否保持稳定。常用的量化指标之一是性能衰减至初始值80%的时间(t 80 ),即当反应持续运行时,活性或法拉第效率下降到初始值80%所经历的时间。该指标可以直观反映催化剂在连续电解条件下的耐久性。在恒电位模式下,通过记录电流密度的变化来判断性能衰减;在恒电流模式下,则通过电位漂移反映反应动力学的稳定性。电化学性能衰减的常见原因包括催化剂溶解(金属离子逐渐进入电解液,使活性位点减少)、颗粒团聚(纳米结构在反应中长大,导致活性表面积缩小)和表面中毒(反应中间体或副产物在表面强吸附,阻碍活性位点)。在工程实践中,常需要结合电解液中金属含量分析、原位表征手段或后表征手段等方式,综合判断衰减的来源与机制。
(2)结构稳定性:从“宏观形貌”到“原子结构”。结构稳定性需通过多种表征手段验证,确保催化剂在反应前后无显著结构变化。
宏观形貌与尺寸:通过透射电子显微镜(Transmission Electron Microscope,TEM)或扫描电子显微镜(Scanning Electron Microscope,SEM)观察催化剂颗粒的尺寸与分散性。例如,反应前Cu基催化剂颗粒的尺寸为5nm,反应后若仍维持在5~8nm,且无明显团聚,则认为其形貌稳定性良好;若颗粒尺寸增加至20nm以上,则会导致活性位点减少、性能衰减。
晶体结构与活性位点:通过X射线衍射(X-ray Diffraction,XRD)分析晶相变化,通过X射线光电子能谱(X-ray Photoelectron Spectroscopy,XPS)分析表面元素价态。例如,Ni基催化剂在反应前为Ni0 /Ni2+ 混合价态,反应后未出现Ni3+ 且Ni0 占比无显著下降,则说明其活性位点结构稳定;若Ni0 完全被氧化为Ni2+ /Ni3+ ,则会导致催化剂活性丧失。
(3)指标间的协同关系与综合评估。电化学碳循环反应的性能指标并非相互独立,它们之间往往存在协同或权衡关系。
协同关系:高法拉第效率与高电流密度同时满足,能够兼顾产物选择性与产能,是工程化应用最理想的状态。
权衡关系:提升电流密度可能导致过电位上升、能耗增加,或降低产物选择性;降低过电位可能牺牲部分速率。因此,实际应用中常需要在能效、产物选择性与速率之间找到平衡点。为避免单一指标造成误解,工程实践中更倾向于采用综合评价方式。例如,通过将能效、法拉第效率与稳定性结合,构建用于比较不同催化体系整体表现的综合评分框架。
综上,法拉第效率、电流密度、过电位与稳定性共同构成电化学碳循环反应的“四维评估体系”。只有在这4个维度均达到行业标准,才能推动技术从实验室走向工业化,为未来能源体系提供核心支撑。
1.2 CO2 还原反应:产物路径与调控CO2 RR是CO2 转化为高附加值碳基燃料和化学品的有效路径,包括C1 产物路径(2~8个电子转移,路径相对简单)和C2 /C2+ 产物路径(不少于12个电子转移,依赖C—C偶联,路径复杂)。产物路径的多样性使产物调控受多因素影响,主要产物路径如图1-3所示[19] 。不同产物路径间竞争的本质是中间体吸附强度与电子转移效率的平衡,这些路径的多样性为CO2 的高值化利用提供了丰富选择,可通过“催化剂设计+反应微环境调控+智能化工艺优化”实现特定产物的定向转化。
(1)C1 产物路径。常见的C1 产物有CO、HCOOH、CH4 等。基于CO2 在催化剂表面的初始质子化步骤,反应路径可划分为两类。一类是羧基路径,即CO2 先转化为中间体*COOH,再经过后续反应生成最终产物,如进一步还原生成CO;另一类是甲酰基路径,CO2 先转化为中间体*OCHO,再通过一系列基元反应生成相应产物,如转化为HCOOH。
图1-3 CO2 RR的主要产物路径[19]
(2)C2 /C2+ 产物路径:常见的C2 /C2+ 产物有C2 H4 、CH3 COOH、C2 H5 OH等。CO2 通过两个*CO[或*CO与其他中间体(如*CH、*CH2 )]的C—C偶联而还原生成C2 产物(甚至是C2+ 产物)。根据目标产物的不同,反应路径可分为生成C2 H5 OH的烷基醇化路径和生成C2 H4 的烷基化路径等。例如,在生成C2 H5 OH的路径中,会涉及中间体*OCHCH2 ,若该中间体继续氢化则可生成C2 H5 OH;而在生成C2 H4 的路径中,*OCHCH2 可能会发生O—C断裂,进而生成C2 H4 。
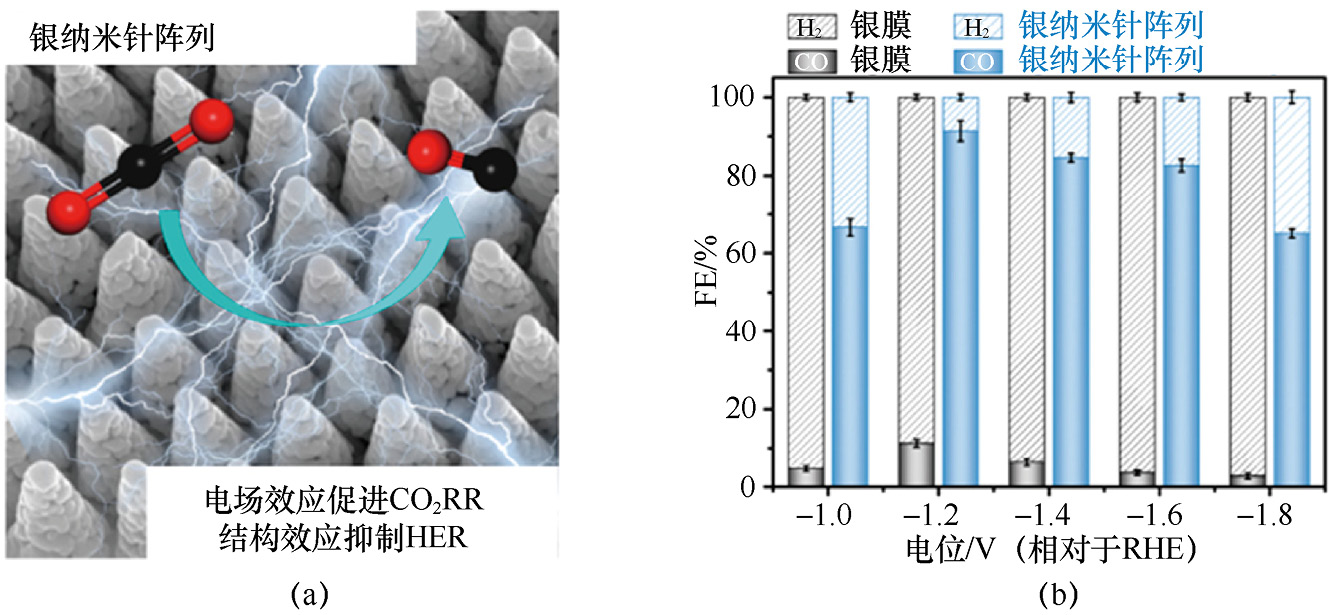
CO2 RR产物调控的核心是通过多维度协同优化,定向强化目标产物的生成路径、抑制竞争反应(如HER、非目标碳产物生成)。CO2 RR产物调控的本质是通过多维度(催化剂结构、电解液环境、外部反应条件、界面状态)协同优化,精准调控中间体吸附、电子转移与C—C偶联等关键过程,最终实现目标产物(C1 或C2 /C2+ )的高选择性生成。例如,通过精心构建有序的银纳米针阵列结构(见图1-4),凭借该结构独特的几何形貌特征,成功实现了材料表面的高疏水性。这种高疏水性能够动态抑制竞争性的HER,进而使得在相对于RHE的电位为-1.0V的条件下,CO的法拉第效率在超过700min的时间里稳定保持在91.4%[20] 。
图1-4 银纳米针阵列结构示意图及其对应的CO2 RR性能[20]
(a)银纳米针阵列结构示意图;(b)对应的CO2 RR性能
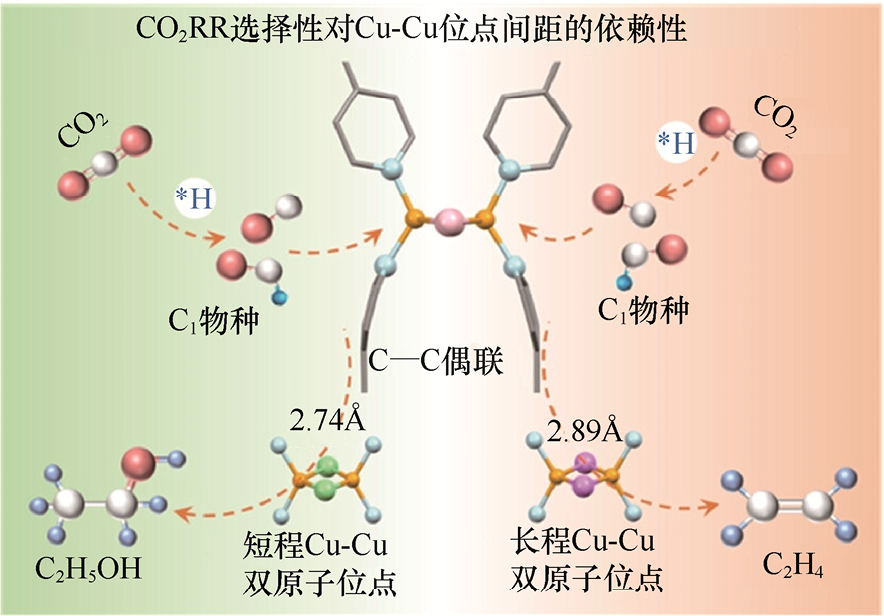
不同金属在CO2 RR中具有各自的本征产物选择性。例如,Au、Ag更易生成CO,Cu是少数能够高效生成C2+ 产物的金属,Sn、Bi则主要生成HCOOH类产物。通过合金化可以进一步调控金属表面对关键中间体的吸附行为,从而改变产物选择性。例如,通过调节Cu与Ag的比例来优化CO覆盖度,可增强后续的C—C偶联步骤,从而提高C2 H4 的产物选择性[21] 。除了金属本身,反应环境也会深刻影响催化路径。电解液组分能够改变界面的微环境与质子传输特性。例如,强碱性电解液有助于抑制HER,但可能引发碳酸盐沉积,从而影响气体传输;较大的阳离子(如K⁺、Cs⁺)可增强界面电场,促进中间体*CO在催化剂表面的积累与相互作用,降低C—C偶联能垒,利于C2+ 产物生成。与此相对应,卤素阴离子不仅能参与界面微环境调节,还能直接影响催化剂表面的原子级构型。如图1-5所示,通过调控卤素对Cu的第一配位壳层作用,可以调整原子级Cu-Cu位点的间距,进而改变*CO的吸附强度及其在表面上的排列方式[22] 。研究结果表明,长程铜–铜(Cu-I-Cu)双位点可加速C2 H4 的生成,而短程铜–铜(Cu-Cl-Cu)双位点则有利于C2 H5 OH的生成,这为理解位点间距依赖的催化性能提供了广泛的认识。
图1-5 第一壳层卤素配位调控原子级Cu-Cu位点间距示意图[22]
1.2.1 CO2 至CO的定向转化 CO2 至CO的定向转化是CO2 利用领域的研究热点之一,目前主要通过电催化、热催化以及等离子体辅助等方法实现。CO2 RR制CO的核心是通过催化剂设计调控活性位点电子结构与中间体吸附强度,抑制HER和C—C偶联等副反应。截至本书成稿之时,针对此类反应的常见催化剂有3类:单原子合金催化剂、双金属催化剂,以及三组分单原子合金催化剂。
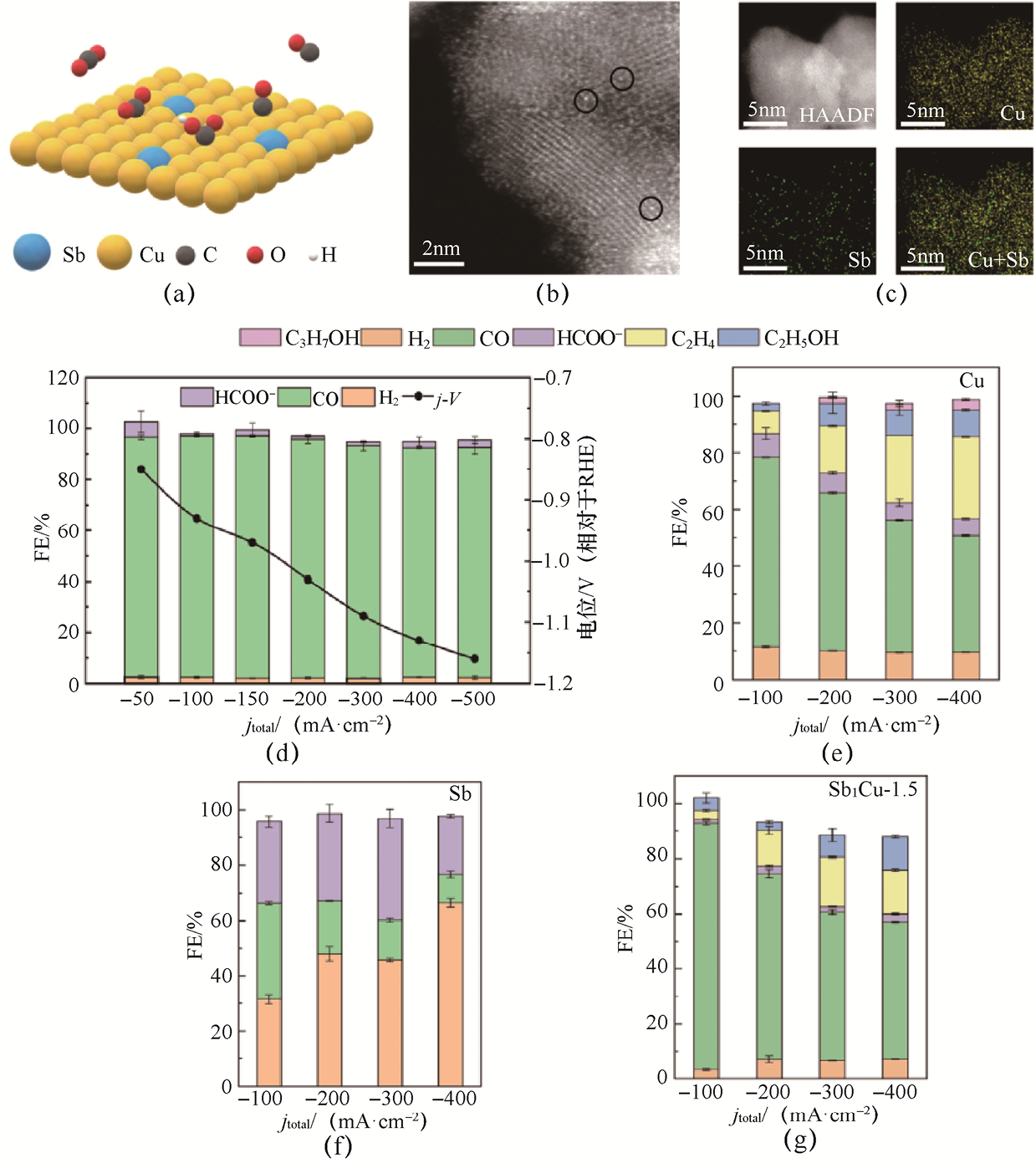
研究表明,在Cu中引入孤立的Sb原子能够显著提升CO2 RR制CO的产物选择性[23] 。通过Sb掺杂铜单原子合金(Sb1 Cu),可在Cu基催化剂中形成稀疏而均匀的单原子掺杂结构,实现对局域电子环境的精准调控。在结构表征中,Sb原子以原子级间隔嵌入Cu晶格(见图1-6),既可保持Cu的连续导电网络,又可避免形成Sb富集相,为后续反应提供电子结构均一的活性界面。电化学测试显示,Sb1 Cu-5的CO2 RR性能显著优于纯Cu和纯Sb催化剂(见图1-6)。例如,在-150mA·cm-2 时,FECO 达到约 95%;即使将部分电流密度提高到-500mA·cm-2 ,FECO 仍能保持在90%以上。Sb1 Cu-5的CO部分电流密度(j CO )最大可达到-452mA·cm-2 ,远超大多数之前报道的非贵金属催化剂。随着电流密度的上升,纯Cu的C2+ 产物比例显著提高;而纯Sb的CO产物选择性很小(FECO 最大值仅约34.8%)。对于Sb1 Cu-1.5,虽然在-100mA·cm-2 时FECO 达到约89%,但在高电流密度下仍有较多C2+ 产物生成。相比之下,Sb1 Cu-5在大电流密度下近乎不生成C2+ 产物。这一系列性能数据直接表明:通过孤立Sb-Cu原子界面的设计,可大幅抑制C—C偶联路径,将产物选择性转向CO,同时保持高电流密度与高催化活性。
图1-6 Sb掺杂的Cu基合金催化剂的结构特征与CO2 RR性能[23]
(a)Sb1 Cu-5催化剂上CO2 转化为CO的示意图;(b)Sb1 Cu-5催化剂的TEM图像,黑色圆圈表示单个Sb原子;(c)Sb1 Cu-5催化剂中Cu和Sb的元素分布;(d)Sb1 Cu-5催化剂的CO2 RR性能;(e)纯Cu催化剂的CO2 RR性能;(f)纯Sb催化剂的CO2 RR性能;(g)Sb1 Cu-1.5催化剂的CO2 RR性能
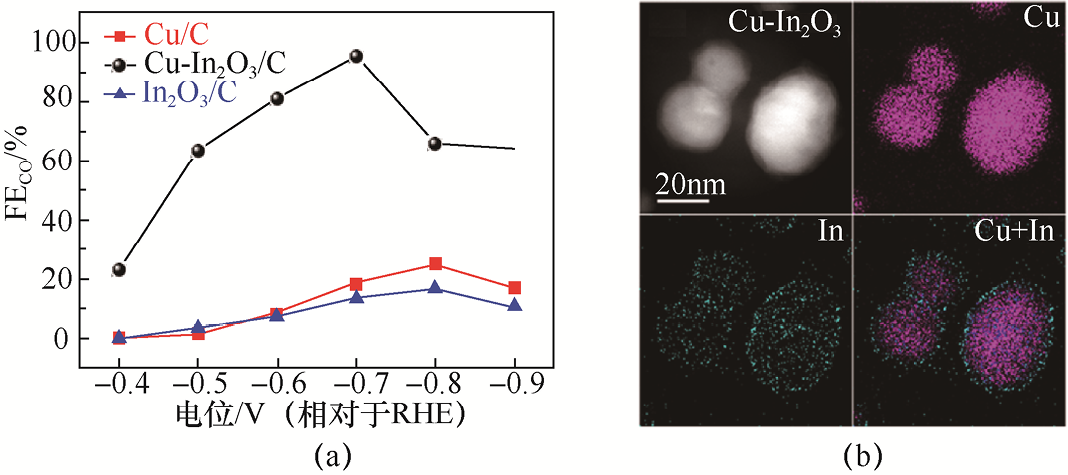
Jia等[24] 提出了一种通过原位电还原铟(In)涂覆CuO纳米线构筑的Cu-In2 O3 /C复合催化剂,用于高产物选择性CO2 RR制CO。结构表征与原位分析表明,在反应过程中,表面的Cu-In物种被逐步重构为In包覆Cu核的核壳结构,从而在界面处形成稳定的电荷转移通道(见图1-7)。电化学测试结合在线微型气相色谱分析显示,Cu/C与In2 O3 /C对照样品的CO产物选择性较低,主要生成H2 或HCOOH;相比之下,Cu-In2 O3 /C复合催化剂在宽电位范围内表现出显著增强的CO生成能力,其FECO 最高可达约95%(见图1-7)。该研究表明,通过构筑稳定的Cu-In异质界面并调控界面电子结构,可有效抑制副反应并引导反应路径向CO定向转化,为双金属催化剂的界面工程设计提供了重要思路。
图1-7 Cu-In2 O3 /C复合催化剂的CO2 RR性能及结构特征[24]
(a)Cu/C、Cu-In2 O3 /C和In2 O3 /C催化剂在不同电位下的CO法拉第效率对比;(b)Cu-In2 O3 催化剂的TEM图像及对应的元素分布
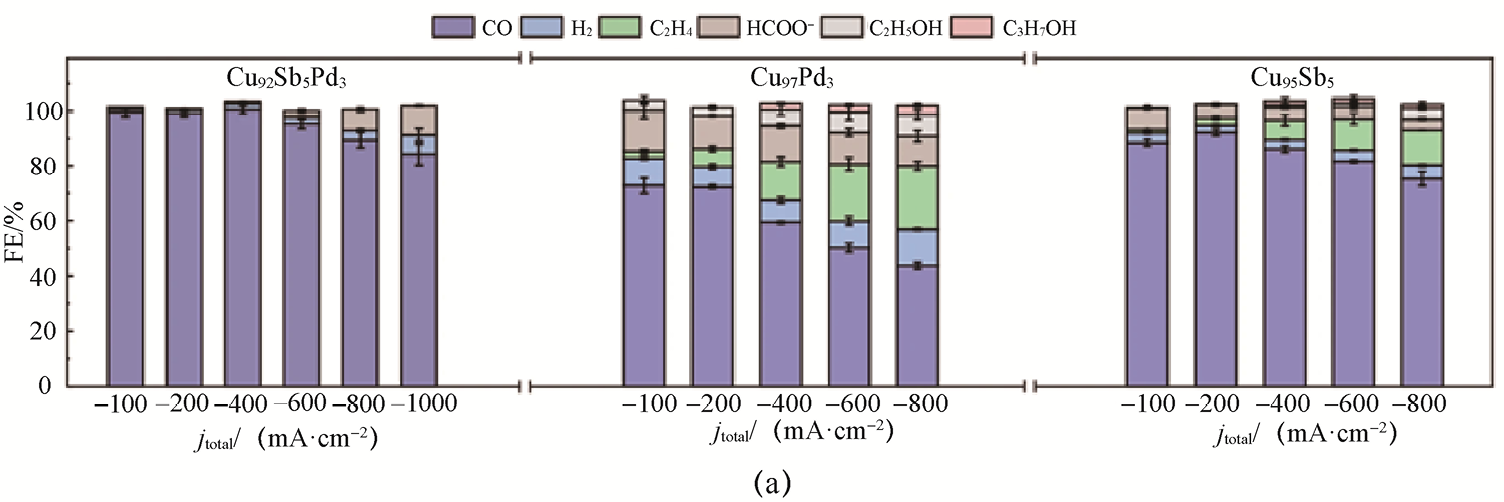
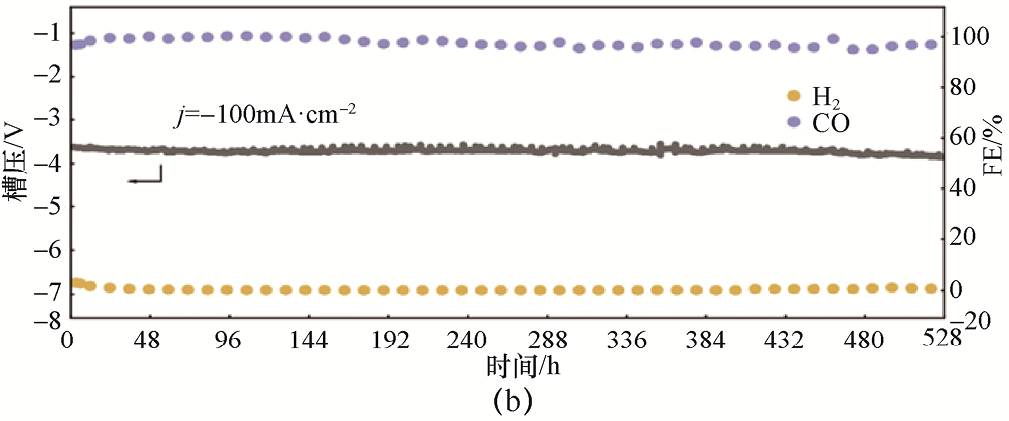
此外,Xue等[25] 提出了一种三组分单原子合金催化剂Cu92 Sb5 Pd3 ,用于高效稳定地通过CO2 RR制CO。该催化剂通过将Sb与Pd单原子协同掺入Cu基体,实现了对Cu局域电子结构的精细调控,从而显著改变了反应路径。电化学性能对比表明,与纯Cu相比,单掺Sb或Pd的Cu97 Pd3 和Cu95 Sb5 在CO产物选择性方面有所提升,但在高电流密度条件下仍会生成C2 H4 、醇类等C2+ 产物,同时HER逐渐占据主导,限制了CO的部分电流密度增长(见图1-8)。相比之下,Cu92 Sb5 Pd3 在宽电位和电流范围内均表现出对CO的高产物选择性。在膜电极组件(Membrane Electrode Assembly,MEA)中,该催化剂在-100mA·cm-2 条件下连续运行528h后,FECO 仍稳定保持在95%以上(见图1-8),展现出优异的长期稳定性。该研究表明,多异原子单原子协同调控是一种将Cu基催化剂转化为高效CO生成电催化反应催化剂(简称电催化剂)的有效策略,为非贵金属体系中实现高产物选择性CO2 →CO提供了重要思路。
图1-8 三组分单原子合金催化剂Cu92 Sb5 Pd3 的CO2 RR性能[25]
(a)Cu92 Sb5 Pd3 、Cu97 Pd3 和Cu95 Sb5 催化剂在不同电流密度下各产物的法拉第效率;(b)Cu92 Sb5 Pd3 催化剂在MEA中的稳定性测试结果
1.2.2 CO2 至羧酸类产物的定向转化 CO2 RR至羧酸类产物的定向转化,核心是通过调控电催化体系、电解质环境、反应电位等关键参数,引导CO2 分子发生特定电子转移与化学键重构,生成HCOOH/HCOO- 、CH3 COOH、草酸(HOOC—COOH)等羧酸产物。根据目标羧酸产物的碳数(C1 、C2+ )和反应路径差异,主要转化方法可分为以下两类。
1.C1 羧酸:HCOOH的定向转化 HCOOH是CO2 RR中最易生成的C1 羧酸产物(二电子转移过程:CO2 +2H+ +2e- →HCOOH),因其反应路径简单、产物选择性易调控,是目前研究最成熟的方向之一。核心在于选择能抑制HER、促进CO2 质子化的催化剂。
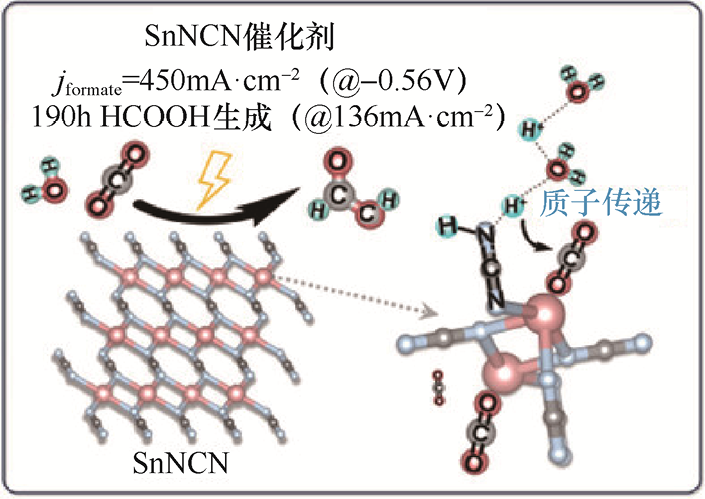
Sn基催化剂是CO2 RR生成HCOOH的经典体系,尤其在中性和碱性电解质中表现出较高的产物选择性。通过调控Sn的表面氧化态,可显著影响反应路径,其中Sn2+ 位点通常被认为是促进中间体*OCHO生成的关键活性中心。针对Sn基催化剂在高电流密度条件下质子传输受限的问题,Zhu 等[26] 提出了一种SnNCN催化剂,在Sn活性中心附近引入[NCN]2- 基团作为内置质子中继站,工作机理如图1-9所示。[NCN]2- 可在N==C==N2- 与N≡≡C—N2- 构型之间发生可逆转变,实现界面质子的快速捕获与释放,从而显著加速质子向中间体*CO2 的定向转移。这种协同机制有效降低了*OCHO的生成能垒,使反应在较低过电位下即可实现高效HCOOH生成。得益于该质子中继效应,SnNCN在较低过电位条件下即可实现工业级电流密度,同时保持超高的HCOOH法拉第效率,并在长时间运行中表现出优异的稳定性。更重要的是,该体系在固态电解质(Solid-state Electrolyte,SSE)反应器中可直接获得高浓度HCOOH产物,避免了传统液相体系中复杂的分离过程,为CO2 RR制HCOOH的工程化应用提供了新思路。
图1-9 在Sn活性中心附近引入[NCN]2- 基团作为质子中继站以促进CO2 RR生成HCOOH的工作机理示意图[26]
铅(Pb)催化剂在酸性电解质中对HCOOH的产物选择性高(>85%),但存在毒性强和导电性差的问题,目前多通过“Pb单原子负载于碳载体”(如Pb-N-C)改善导电性和原子利用率,降低毒性风险。铋(Bi)是环境友好型催化剂,通过电沉积制备的Bi纳米片或Bi2 O3 衍生催化剂,在中性电解质中(如KHCO3 溶液)可实现对HCOOH的高产物选择性(>95%),且能抑制CO等副产物生成,适合低能耗场景。通过结构设计,Bi基催化剂的HCOOH产物选择性普遍超过90%。例如,Chen等[27] 开发的Bi2 O2 NCN衍生催化剂,在-0.6V(相对于RHE)下法拉第效率达98.3%;Zhao等[28] 制备的Bi/BiOBr-D催化剂在流动电解槽中实现了90.6%的法拉第效率,分电流密度高达815mA·cm-2 。
2.C2+ 羧酸:乙酸、丙酸等的定向转化 C2+ 羧酸(如CH3 COOH)需通过多电子转移和C—C偶联反应(如2CO2 +8H+ +8e- →CH3 COOH+2H2 O)实现,反应路径更复杂,核心挑战是抑制C1 产物(CO、HCOOH)并促进关键C—C偶联步骤。截至本书成稿之时,这类反应仍主要依赖Cu基催化体系,但裸Cu催化剂往往产物分散,易生成CO、H2 及醇类副产物,亟待通过结构与反应微环境调控提升羧酸产物选择性。近期,有研究提出“自增压纳米胶囊”策略,通过在Cu催化位点外构筑限域壳层,实现对局域反应环境的精准调控[29] 。如图1-10所示,该纳米胶囊结构在反应过程中可在内部形成局域CO富集和轻微增压效应,从而显著提高*CO覆盖度并延长其停留时间,促进CO—CO偶联及后续含氧C2+ 中间体的生成。在此基础上,反应路径可在CH3 COOH或C3 H7 OH之间切换,取决于局域质子供给与中间体加氢程度。电化学测试表明,该催化剂在中性条件下可实现对CH3 COOH或C3 H7 OH的高定向转化,其中CH3 COOH(或C3 H7 OH)的法拉第效率可超过60%,对应的部分电流密度达到200mA·cm-2 以上,并在长时间运行中保持稳定。这一工作表明,通过“纳米限域+自调控反应压力”的方式,可在不依赖复杂合金化的前提下有效调控C2+ 羧酸转化路径。
除了通过调控催化剂本身的结构与活性来提升性能,还可以通过优化电解质成分和反应条件来促进CH3 COOH的高效生成。高浓度碱性电解液有助于CO2 在界面生成4 ),这类溶液可降低CO2 RR的过电位,同时稳定关键中间体*CO,从而增强C2+ 羧酸的产物选择性。
图1-10 自增压纳米胶囊催化剂促进CO2 RR生成CH3 COOH/C3 H7 OH的机理示意图[29]
1.2.3 CO2 至烃类产物的定向转化 CO2 至烃类产物[如CH4 、C2 H4 、丙烷(C3 H8 )等]的定向转化是“碳捕集、利用与封存”体系的核心技术,可将温室气体转化为高能量密度的燃料或化工原料,实现“碳循环”闭环。该过程的核心挑战是抑制HER、CO等副反应,同时通过精准调控催化体系与反应条件,促进C—C偶联与深度加氢,最终定向生成目标烃类。下面从反应基础原理、核心调控要素、典型催化体系,以及现存挑战与未来发展方向这4个方面展开介绍。
1.反应基础原理:烃类产物的生成路径与关键中间体 CO2 至烃类产物的定向转化是多电子、多步骤的复杂还原过程(需8~18个电子转移),核心是“C1 中间体生成→中间体转化与烃类生成”两步,关键中间体的竞争反应直接决定产物选择性。CO2 定向转化为烃类产物的核心反应路径如下。
(1)C1 中间体生成(CO2 活化)。CO2 在阴极催化剂表面首先接受电子与质子,生成两种关键中间体,即羧基中间体(*COOH)和甲酰基中间体(*OCHO),二者的竞争决定后续产物分支。
CO2 +e- +H+ →*COOH
CO2 +e- +H+ →*OCHO
*COOH的生成是主流路径,可进一步生成烃类和CO;*OCHO的生成是副路径,易进一步加氢生成甲酸盐HCOO- ,需抑制。
(2)中间体转化与烃类生成。*COOH进一步反应生成羰基中间体*CO(*COOH→*CO+H2 O)。*CO是生成烃类的关键中间体,其后续反应分为以下两类。
生成C1 烃(CH4 ):*CO需经历“深度加氢”,路径为*CO→*CHO→*CH2 O→*CH3 O→CH4 (需抑制C—O偶联,强化加氢活性)。
生成C2+ 烃(C2 H4 、C3 H8 ):*CO需先经历C—C偶联,再通过加氢生成多碳产物,典型路径(如C2 H4 )为2CO→*CO—CO(二聚中间体)→*CH2 —CH2 →C2 H4 (需提高CO覆盖度,降低偶联能垒)。
2.核心调控要素:决定烃类产物选择性的三大关键变量 CO2 RR生成烃类的产物选择性并非由单一因素决定,而是“催化剂-电解液-反应条件”协同作用的结果。
催化剂是产物选择性的核心调控单元,通过影响CO2 的吸附方式以及关键中间体(*COOH、*CO、*CHx 2+ 烃类的金属主要是Cu,它具有独特的*CO吸附强度和表面反应特性,在多碳产物生成中具有不可替代性。相比之下,其他金属更倾向于生成CO或发生HER,难以实现C—C偶联。对Cu基催化剂的调控主要集中在晶面取向、尺寸和形貌等方面。不同晶面对中间体的吸附行为存在显著差异,会导致产物分布不同。例如,某些低指数晶面更有利于*CO的深度加氢,而富含方形对称位点的晶面可增强*CO吸附并降低C—C偶联能垒,优先生成C2 烃类;高指数晶面因暴露更多低配位原子位点,往往能够促进更高碳数烃类的生成。此外,纳米结构化设计通过增大比表面积和优化局域传质条件,可显著提高*CO覆盖度,从而提升C2+ 烃的整体产物选择性。进一步地,通过合金化、异原子掺杂或构建异质结,还可调节Cu的电子结构与界面电荷分布,协同增强C—C偶联过程并抑制HER。
电解液在反应中起到“微环境调控者”的作用,通过影响界面环境pH值、质子供给速率及离子特性,可间接调控中间体转化路径。碱性条件通常有利于抑制HER,并促进活化CO2 物种和*CO的表面积累,从而增加C—C偶联概率;相反,强酸性环境中质子浓度过高,易导致HER占优,不利于多碳烃生成。
反应条件则可被视为反应动力学的“加速器”,通过调控电子转移速率和中间体停留时间,可进一步优化产物分布。在过电位较低的条件下,反应动力学受限,中间体更倾向于逐步加氢,生成低碳产物;随着过电位的提高,电子转移加快,*CO覆盖度上升,有利于C—C偶联和多碳烃生成。
此外,温度和压力也会对反应产生显著影响:适度升温可优化反应动力学的性能,但过高温度可能削弱中间体稳定性;提高体系压力则有助于增加CO2 溶解度和界面*CO覆盖度,从而进一步促进C2+ 烃类的生成。
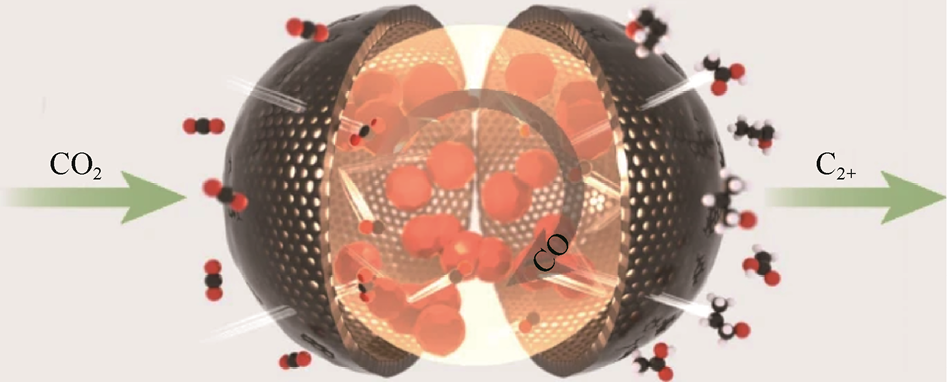
3.典型催化体系:针对不同烃类产物的定向设计 高产物选择性生成C1 烃(CH4 )的催化体系的核心设计逻辑在于抑制C—C偶联并强化*CO的深度加氢路径。相关研究表明,通过引入非金属掺杂调控Cu的局域电子结构,可有效引导反应向CH4 分支发展。以N掺杂碳负载的Cu纳米颗粒催化剂为例,该催化剂中N原子与Cu形成稳定的配位作用,显著调节了Cu的d带中心,使*COOH→*CO→*CHO的逐步加氢过程在反应稳定性和速率方面都更优[30] 。电化学测试显示,该催化剂在1mol·L-1 KOH电解液中于过电位较低的条件下实现了最高约73.4%的CH4 法拉第效率,明显优于未掺杂碳载Cu对照样品,其产物分布中C2+ 烃与含氧产物均受到有效抑制。原位与理论分析进一步表明,N掺杂位点不仅增强了中间体*CHO的稳定性,还削弱了CO—CO偶联倾向,从而在反应路径层面强化了C1 产物选择性。高产物选择性生成C2+ 烃(C2 H4 、C3 H8 )体系则需要增强*CO覆盖度,降低C—C偶联能垒。如Cu-Ag合金体系中,通过精确调控双金属组成,可显著提升C2 H4 的产物选择性[31] 。以Cu67 Ag33 纳米复合催化剂为例,该材料的前驱体在反应过程中经历由液–液界面诱导的原位重构,形成Ag富集表层与Cu富集次表层协同分布的异质结构,促进C2 H4 的高效合成。Cu67 Ag33 纳米复合催化剂上CO2 RR的机理与性能(见图1-11)表明,Ag位点在较低过电位下优先催化CO2 向CO的转化,生成的*CO随后在界面处发生横向迁移并富集于相邻的Cu位点,从而在Cu表面形成高*CO覆盖度环境。这一CO溢流(CO Spillover)效应显著激活了CO—CO偶联路径,降低了C—C偶联能垒,最终提高了C2 H4 产物选择性。同时,液–液界面诱导的结构可重构Cu位点,保持适中的CO吸附强度,避免*CO过度解吸或被深度加氢为CH4 ,从反应路径层面实现了对C2 H4 生成分支的精准调控。
图1-11 Cu67 Ag33 纳米复合催化剂上CO2 RR的机理与性能[31]
(a)Cu67 Ag33 催化剂上CO2 RR的机理示意图;(b)Cu67 Ag33 催化剂上CO2 RR的性能
4.现存挑战与未来发展方向 截至本书成稿之时,CO2 至烃类的定向转化仍面临活性低、产物选择性易衰减、能耗高这三大核心挑战,未来需从以下方向突破。
(1)高效催化剂精准设计。结合原位表征[如原位拉曼光谱(Raman Spectrum)、原位XPS]与密度泛函理论(Density Functional Theory,DFT)计算,揭示真实反应中间体(如*CO—CO、*CH2 —CH2 ),开发单原子Cu基催化剂、高指数晶面Cu催化剂等,实现中间体吸附能的定量调控。
(2)电解液与反应器创新。设计功能化离子液体电解液,同时提升CO2 溶解度与质子传输效率;开发流动电解槽,替代传统H形电解槽,增强CO2 传质,推动电流密度从“百毫安每平方厘米级”迈向“1A·cm-2 级”(降低工业化能耗)。
(3)稳定性提升策略。通过表面包覆(如N掺杂碳层)、载体改性(如TiO2 @C核壳结构),抑制Cu催化剂的氧化与团聚,实现超长时间的稳定运行(满足工业化寿命需求)。
(4)产物分离与系统集成。开发“电催化-产物分离”一体化装置,解决烃类产物在电解液中的溶解与分离难题,降低后续提纯成本。
综上所述,CO2 至烃类的定向转化是一项多学科交叉的技术,其突破不仅依赖催化材料的创新,还需电解液、反应器与系统集成的协同优化,最终实现从实验室研究到工业化应用的跨越。
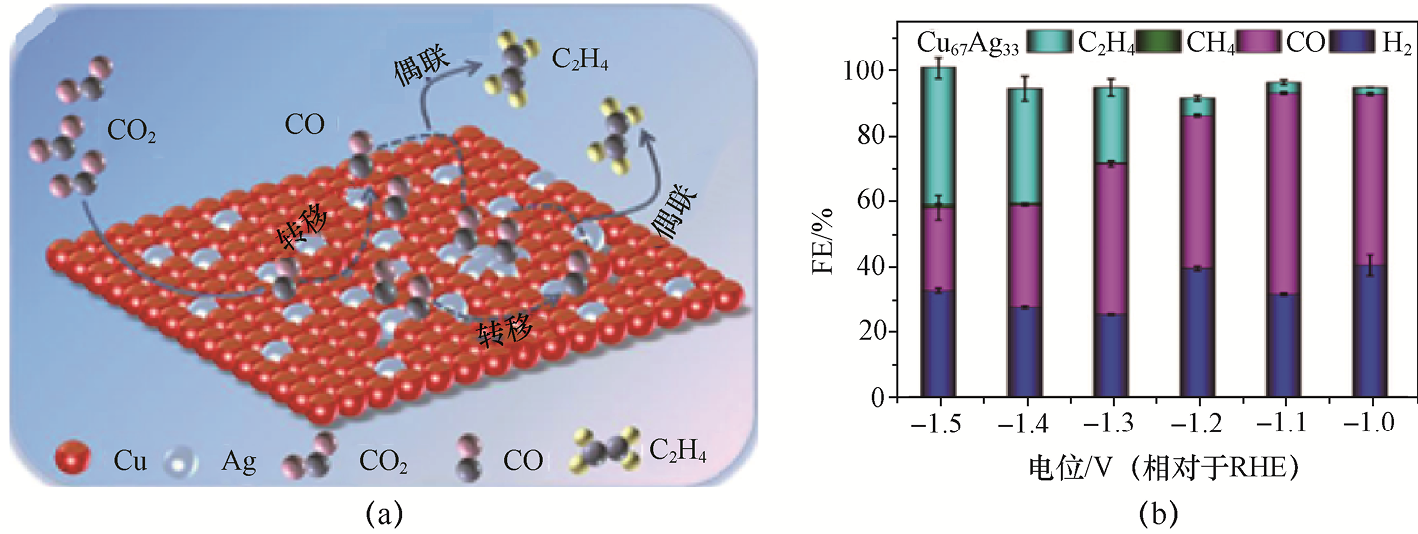
1.2.4 CO2 至醇类产物的定向转化 CO2 至醇类产物的定向转化是实现碳减排与高附加值碳资源利用的重要研究方向。中国科学院化学研究所团队[32] 采用分步沉淀与分步煅烧相结合的策略,提出了具有明确界面结构的Pr-Cu氧化物异质界面催化剂(Pr6 O11 -Cu-SS)。如图1-12所示,该催化剂中Pr6 O11 与Cu之间通过Pr—O—Cu紧密耦合,在反应界面形成一种“活性锁定”结构,使Cu在电解过程中稳定维持Cuδ+ /Cu0 混合价态,并在局部电场与电子转移驱动下实现动态自调节,构建出具有“自修复特征”的稳定异质界面。这种界面结构一方面增强了CO2 的活化能力,促进*CO的持续生成与富集;另一方面通过稳定*CHO、*CH2 CHO等关键中间体,有效抑制C2 H4 生成路径,显著推动C—C偶联后向醇类产物的深度氢化。在-1.08V(相对于RHE)条件下,该催化剂实现了700mA·cm-2 的高电流密度,对应C2+ 醇的总法拉第效率达71.3%,其中C2 H5 OH和n -C3 H7 OH的法拉第效率分别为58.6%和12.7%,C2+ 醇与C2 H4 的生成比例高达12∶1。该工作表明,通过异质界面“价态锚定–反应路径引导”的协同设计,可在高电流密度条件下实现CO2 向高碳醇类的高效、稳定转化。
图1-12 Pr6 O11 -Cu-SS的CO2 RR机理与性能[32]
(a)利用Pr6 O11 -Cu-SS的CO2 RR制C2+ 醇的机理;(b)Pr6 O11 -Cu-SS的CO2 RR性能;(c)Pr6 O11 、Pr6 O11 -Cu-CS、Pr6 O11 -Cu-SS、Pr6 O11 -Cu-SC和CuO在700mA·cm-2 电流密度下产物的法拉第效率
此外,韩国光州科学技术研究院团队[33] 将磷化铜(CuP2 )与镍铁(NiFe)氧化物催化剂整合到MEA中,制备出一种富磷铜催化剂。该催化剂在电化学装置中,C3+ 产物的法拉第效率达到66.9%,部分电流密度为-735.4mA·cm-2 ,生成速率为1643μmol·cm-2 ·h-1 ,电极单位面积可施加-1100mA·cm-2 的电流。他们揭示了一条新的反应路径,即在甲酸酯转化为HCHO中间体基团的过程中形成C—C,从而将CO2 转化为烯丙醇。美茵茨约翰内斯·古腾堡大学的卡斯滕·施特雷布(Carsten Streb)教授团队[34] 设计了一种专门涂有钴铜粉末的电极,可先使CO2 在钴(Co)的作用下分解产生CO,再令Cu催化CO,转化为C2 H5 OH。该方法将CO2 转化为C2 H5 OH的产物选择性超过70%。
1.2.5 CO2 至醛类产物的定向转化 CO2 分子具有稳定的线性结构,需通过电子转移+质子耦合打破化学键,逐步还原为醛类。该定向转化核心是CO2 先在阴极催化剂表面接受电子,生成关键中间体,再经选择性质子化,生成醛基(—CHO),避免被过度还原为醇、烷烃或完全还原为CH4 。以生成CH3 CHO为例,典型反应路径如下(酸性/中性条件)。
第一步是CO2 活化与中间体生成。首先,CO2 +e- →CO2 - ·;然后,+ →HCOO·(甲酸基自由基)或+ →CO+OH- (若生成CO中间体,需进一步加氢)。
第二步是C—C偶联与碳链增长。若以CO为中间体,2个*CO先在催化剂表面偶联生成*OC—CO(二羰基中间体),再接受电子+质子,生成HOOC—CHO(乙醛酸)。
第三步是选择性还原为醛基:2HOOC—CHO+2e- +2H+ →CH3 CHO+2CO2 +H2 O(或直接质子化,生成CH3 CHO,避免加氢为C2 H5 OH)。总反应(以CH3 CHO为例):2CO2 +10H+ +10e- →CH3 CHO+3H2 O。
醛类的生成效率和产物选择性,非常依赖催化剂的电子结构调控与反应环境的匹配,需抑制HER、CO生成、过度还原(如醇、烷烃)等副反应。截至本书成稿之时,CO2 至醛类产物的定向转化仍面临三大核心挑战,它们也是重要的研究方向。
(1)产物选择性与活性的平衡问题。醛类是CO2 RR的中间产物,易进一步加氢为醇(如CH3 CHO→C2 H5 OH)或分解为CO,难以稳定生成,可以通过设计限域活性位点[如金属有机骨架(Metal-organic Framework,MOF)包裹催化剂],通过空间位阻限制中间体的过度加氢。
(2)催化剂长期稳定性问题。金属(如Cu、In)催化剂在电催化过程中易发生溶解、团聚或氧化(如Cu→Cu2+ ),导致活性下降。可通过载体修饰、表面包覆等提升催化剂活性。
(3)规模化应用的成本与设备适配问题。单原子催化剂(Single-atom Catalyst,SAC,如Ni-N-C)的制备成本高,且现有电解槽(如H形电解槽)的电流密度小,难以满足工业化需求。因此,需要开发低成本催化剂(如用Cu基合金替代单原子催化剂),并采用流动电解槽(而非H形电解槽),以提高CO2 传质效率,提升电流密度,为工业化奠定基础。
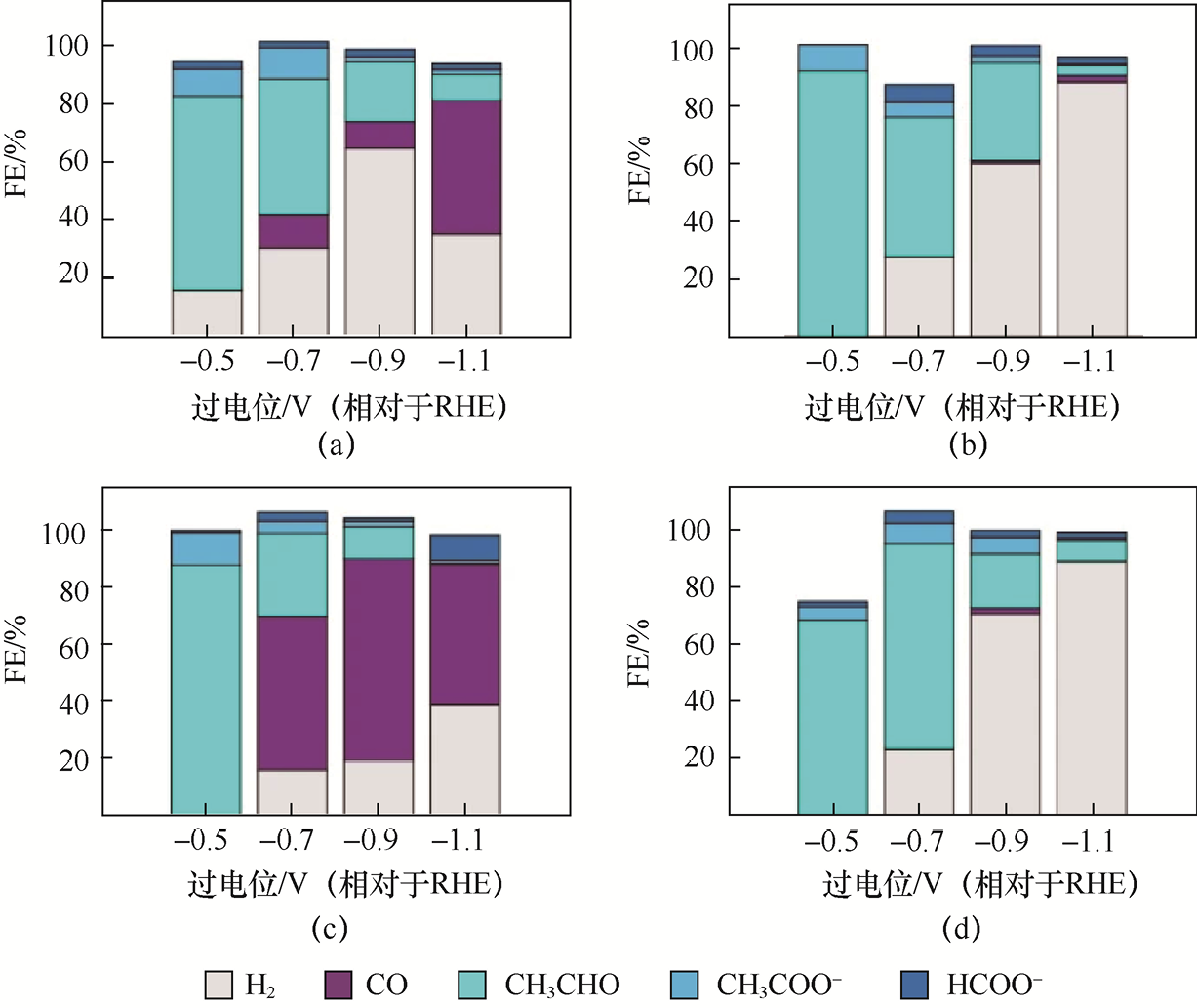
例如,Koolen等[35] 提出了一系列通过火花消融法制备的约1.6nm Cu簇催化剂,并分别固定在不同载体上以研究对CO2 RR生成CH3 CHO的法拉第效率(见图1-13)。在0.1mol L-1 KHCO3 电解液中的筛选结果显示:在较低的过电位(约-0.5V,相对于RHE)下,Cu-NCB(N掺杂炭黑载体)催化剂的CH3 CHO法拉第效率约为66%;同条件下,Cu-GO(石墨氧化物载体)催化剂表现出更高的产物选择性,法拉第效率可达约92%;当在Cu簇内引入少量Ag并分别负载于两种载体时,Cu-Ag-NCB催化剂在-0.5V条件下的CH3 CHO法拉第效率高达约88%,而Cu-Ag-GO催化剂在-0.7 V条件下仍可维持约70%的CH3 CHO法拉第效率。这些结果表明,不同载体与簇组成不仅能影响中间体吸附环境,还能调节产物分布和电子结构,使CH3 CHO的生成比在较宽电位范围内得到增强。
CO2 至醛类产物的定向转化,是实现碳资源化的重要路径。该过程的关键在于通过催化剂电子结构调控(如合金化、单原子设计)和反应环境优化(如中性电解质、适度加压)来精确调节中间体(*CO、*HCOO)的选择性质子化路径,从而实现醛类产物的高效生成。截至本书成稿之时,这类技术在实验室条件下已能够实现较高的醛类产物选择性,但在规模化应用中仍面临稳定性、成本及传质效率的挑战。未来,借助材料科学、电化学与化工技术的交叉整合,有望实现从实验室研究到工业化应用的转化,为碳减排和绿色化工提供可行的技术方案。
图1-13 不同的Cu簇催化剂上CO2 RR生成CH3 CHO的法拉第效率[35]
(a)Cu-NCB催化剂;(b)Cu-GO催化剂;(c)Cu-Ag-NCB催化剂;(d)Cu-Ag-GO催化剂
1.3 含碳小分子的电氧化含碳小分子的电氧化通常涉及多电子/多质子耦合转移过程,其反应路径、动力学行为及产物选择性对催化剂表面结构、电子性质及反应界面环境高度敏感。不同含碳小分子在分子结构、键能分布及反应活性上的差异,使其在电氧化过程中呈现出各具特征的反应机理与能量需求。因此,有必要分别围绕典型含碳小分子的电氧化展开讨论,以系统阐明其反应特征与催化调控规律。
1.3.1 CO的电氧化 作为含碳小分子中最基础且活性较高的代表,CO的电氧化不仅在能源转化与存储体系中具有重要意义,还在气体传感以及高值化学品合成等领域发挥着关键作用。CO分子的结构简单,C≡≡O的键能高(1072kJ·mol-1 ),在电极表面的吸附、活化与氧化过程高度依赖电极材料的表面电子结构及反应环境,因此深入理解机理和设计高效催化剂是推动其电氧化技术发展的核心。
1.CO电氧化的基本反应路径 研究表明,Pt基催化剂表面CO的电氧化机理主要遵循朗缪尔-欣谢尔伍德机理(Langmuir-Hinshelwood Mechanism,简称L-H机理),即反应由吸附态的CO分子与吸附态的含氧物种(通常为OH)构成的“反应对”共同参与[36-38] 。根据L-H机理,反应动力学主要取决于Pt表面吸附CO和OH的活性及它们的迁移行为。Pt表面的CO氧化可通过两类动力学模型来描述:平均场近似模型和成核生长模型。二者的核心差异在于吸附态CO分子的迁移速率。平均场近似模型假设吸附态CO分子的迁移速率远大于反应速率,因此反应速率与表面CO和OH的平均浓度成正比;成核生长模型则认为吸附态CO分子的迁移非常缓慢,反应主要发生在CO邻近缺陷位或台阶位的OH上,通过局部生成空位吸附OH,反应才得以推进。不同模型反映了Pt表面结构及CO吸附行为对反应动力学的显著影响。
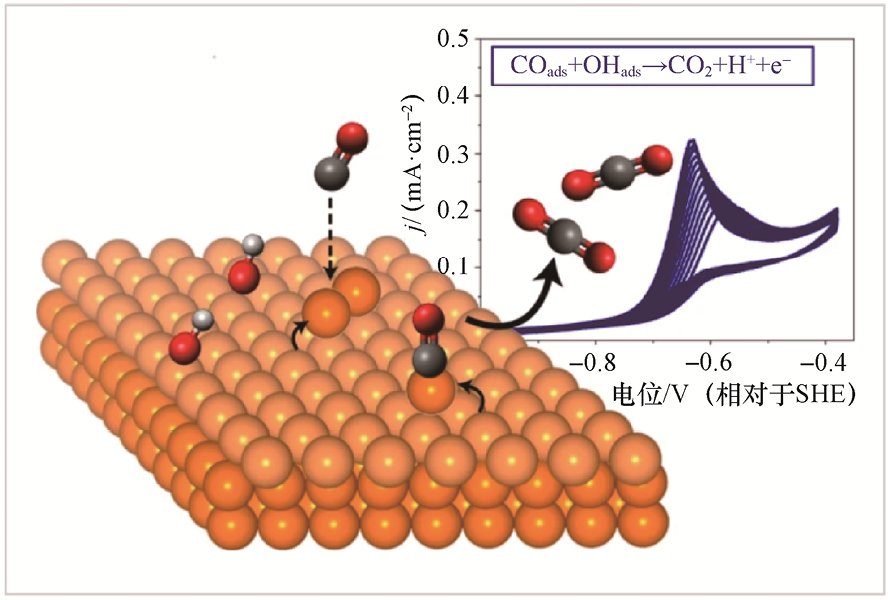
2.CO电氧化的应用 因斯布鲁克大学尤莉娅·孔策-利布豪泽(Julia Kunze-Liebhäuser)团队[39] 系统研究了铜电极在碱性介质中CO电氧化的本征行为,发现单晶Cu(111)表面可在低过电位条件下高效驱动CO电氧化,性能显著优于传统Pt、Au等贵金属催化剂。循环伏安法测定结果显示,仅在CO饱和的电解质中才出现清晰而强烈的氧化峰,明确了CO电氧化相对于标准氢电极(Standard Hydrogen Electrode,SHE)的起始电位(Onset Potential)为-0.73V,对应过电位约为180mV,最大电流密度可达0.35mA·cm-2 。更重要的是,CO的电氧化过程发生在Cu本体氧化之前,避免了不可逆氧化导致的失活,为催化剂在过电位较低时的稳定运行提供了保障。
图1-14直观地展示了这一“自激活”机理:在外加电位作用下,单晶Cu(111)表面并非静态反应位点,而是经历了可逆的电位驱动表面重构,从而原位生成高活性的CO氧化位点。原位电化学扫描隧道显微镜(Scanning Tunneling Microscope,STM)分析进一步揭示了这一过程的微观细节:初始平整的单晶Cu(111)表面在电位升高至约-0.45V(相对于SHE)时发生了明显的重组,形成了线状纳米结构和吸附的Cu原子团簇,这些动态生成的低配位位点与CO氧化活性同步增强;当电位回扫至更低的负区间(约-0.85V,相对于SHE)时,表面结构又完全恢复为平整金属态,证明该重构过程具有高度可逆性。
该研究首次建立了“电位调控-表面自重构-高效CO氧化活性”的直接构效关系,揭示了铜电极在反应条件下通过自适应结构演化实现高活性与稳定性统一的机制,为非贵金属电催化剂在CO电氧化及相关电化学反应中的理性设计提供了重要理论与实验依据。
图1-14 单晶Cu(111)表面在低过电位条件下高效驱动CO电氧化的机理示意图[39]
1.3.2 低碳烃类分子的定向电氧化 CH4 、乙烷(C2 H6 )、C2 H4 等低碳烃类分子由于储量丰富、价格低廉,成为电化学转化过程中的重要原料分子。然而,这类分子的化学惰性强(甲烷C—H约为439kJ·mol-1 ,乙烷中第一个C—H的平均解离能约为423kJ·mol-1 ),同时氧化产物多样、路径复杂,因此在温和条件下实现其高定向电氧化具有极大挑战性。但通过合理设计催化剂的活性位点、调控反应中间体吸附能及反应微环境,可以有效促进低碳烃类分子的活化与定向转化,这已成为电化学转化研究中的重要方向。
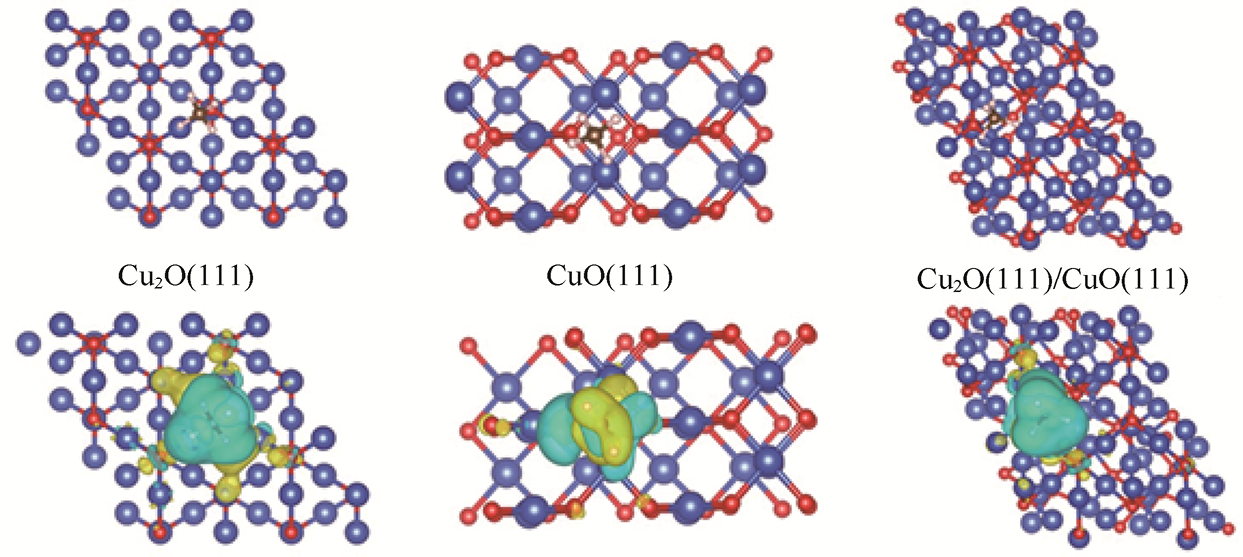
1.CH4 的电氧化 CH4 作为页岩气、沼气等的主要成分,用途极其广泛,在能源领域可以作为燃料,也是重要的化工原料。随着科技发展,CH4 的利用方式也在不断创新,CH4 的电氧化反应的理想产物包括CH3 OH、HCHO和HCOOH等具有高附加值的含氧化学品,因此该反应被认为是推动能源与环境可持续发展的潜在反应路径之一。然而,该反应面临的主要挑战在于阳极析氧反应(Oxygen Evolution Reaction,OER)的竞争以及对目标产物选择性的控制。北京化工大学陈咏梅课题组[40] 研究了Cu2 O/CuO复合材料在电化学CH4 转化制备C2 H5 OH的催化行为,并结合理论与原位表征揭示了反应机理,如图1-15所示。DFT计算表明,在Cu2 O/CuO异质界面上,CH4 及关键中间体(如*CH3 、*CH2 、*CH2 OH)相对于单一CuO或Cu2 O表面具有更强的吸附能和更低的反应能垒,这表明该界面能促进CH4 活化和C—C偶联过程。具体而言,与Cu2 O或CuO单独存在时的较弱吸附相比,Cu2 O/CuO界面能显著增强CH4 的初始吸附与关键中间体*CH2 OH的稳定性,从而为随后与*OH自由基的反应提供有利的动力学条件。结合电化学测试与电子顺磁共振(Electron Paramagnetic Resonance,EPR)分析可进一步证实,在动力学活化过程中,被活化的CH4 可与阳极表面水氧化过程原位生成的羟基自由基(·OH)发生反应,沿着*CH3 →*CH2 →*CH2 OH等路径逐步生成C2 H5 OH。该工作表明,通过构建具有适宜中间体吸附能的Cu2 O/CuO异质界面,不仅可以改善CH4 的C—H断裂与氧化吸附,还将热力学优势转化为可控的反应路径,提高了常温条件下CH4 向C2 H5 OH转化的产物选择性与效率。
图1-15 Cu2 O(111)、CuO(111)和Cu2 O(111)/CuO(111)表面的CH4 吸附及电荷密度差异[40]
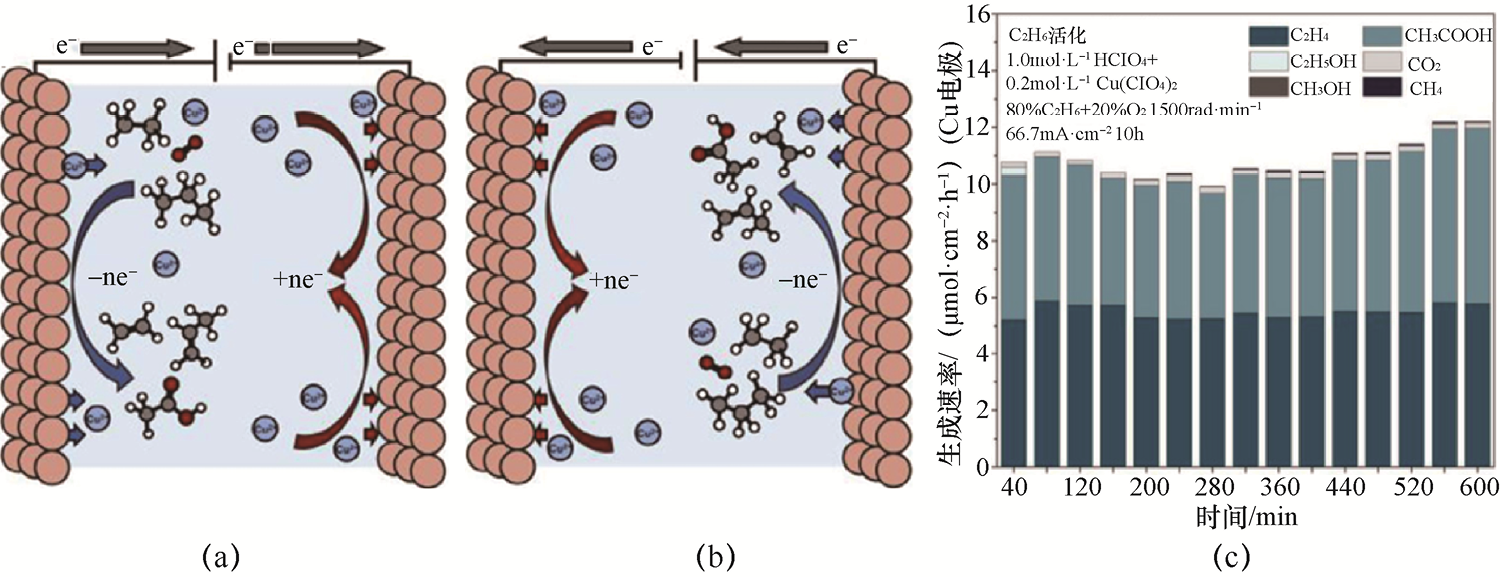
2.C2 H6 与C2 H4 的电氧化 C2 H6 与C2 H4 在石化工业中广泛存在,对其进行电氧化反应可以制备C2 H5 OH、CH3 CHO、CH3 COOH等高附加值含氧有机物,因此该反应是实现能源转型的重要方向之一。C2 H6 中C—H的键能虽略低于CH4 (约423kJ·mol-1 ),但氧化路径复杂,容易生成C—C断裂产物。Cu基电极因其独特的中间体吸附特性,在C2 H6 电氧化中显示出较好的产物选择性。例如,以多晶铜板作为电极,氧气(O2 )为氧化剂,在常温常压下即可高效地将C2 H6 和C3 H8 转化为高附加值的烯烃和含氧化合物[如C2 H4 、CH3 COOH和丙烯(C3 H6 )]。如图1-16所示,在整个实验过程中C2 H4 和CH3 COOH的生成速率保持稳定,每平方厘米Cu电极上的生成速率分别为5.51µmol·h-1 ±0.23µmol·h-1 和5.11µmol·h-1 ±0.51µmol·h-1 ;通过周期性反转电流方向,该系统不仅显著提高了反应速率和稳定性,还实现了Cu电极的循环使用,从而克服了传统热催化方法的高能耗、高排放和催化剂失活等问题,为轻质烷烃的温和转化提供了新策略。
相对来说,对C2 H4 电氧化的研究更加成熟。C2 H4 分子中存在C==C,使其在电极表面易于被吸附并发生加成反应。在贵金属(如Pd、Pt)电极上,C2 H4 易于被氧化为乙二醇[(CH2 OH)2 ]或CH3 CHO。电沉积制备的Pd被认为是极具应用潜力的电催化材料,然而,(CH2 OH)2 氧化反应中的电流密度、法拉第效率和产率较低,限制了其实际应用。清华大学段昊泓、李必杰团队[42] 利用表面离子修饰策略,开发了一种高效的Pd基催化剂,直接通过电化学反应将C2 H4 转化为(CH2 OH)2 。通过在Pd表面引入氯离子(Cl– ),可以显著提高(CH2 OH)2 的电流密度和法拉第效率。具体而言,Cl– 的强电子吸引特性可以调节Pd表面的电子结构,抑制(CH2 OH)2 的过度氧化,并改变(CH2 OH)2 在Pd表面的吸附构型,从而提高产物选择性。
图1-16 C2 H6 与C3 H8 的电氧化实验系统示意图及其产物分布与生成速率[41]
(a)电化学促进的C2 H6 和C3 H8 活化体系示意图,橙色、白色、灰色和红色的球分别表示Cu、H、C和O原子,蓝色球表示Cu离子;(b)通过调节电压极性或反转电流方向实现阳极与阴极切换的示意图,可以防止阳极Cu电极的溶解;(c)产物分布与生成速率
3.催化剂设计策略 针对该类催化剂,研究人员提出了多种创新设计策略,核心目标是在不牺牲催化活性和稳定性的前提下显著降低贵金属(如Pt)的用量,从而有效节约催化剂制备成本。典型策略主要包括:合金结构低Pt电催化剂、核壳或空心结构低Pt电催化剂,以及基于多尺寸协同效应的低Pt电催化剂。其中,合金结构低Pt电催化剂通过精确调控Pt与其他金属的合金化程度及电子结构,实现对催化活性和稳定性的同步提升。核壳或空心结构低Pt电催化剂则依托独特的结构设计,使Pt原子或纳米颗粒高度分散于表面或壳层中,从而在显著降低Pt用量的同时最大化其原子利用效率。相比之下,多尺寸协同效应低Pt电催化剂通过不同尺寸Pt物种之间的协同作用,优化反应物吸附与反应动力学过程,从而进一步提升整体电催化性能。
1.3.3 醇类分子的电氧化 CH3 OH、C2 H5 OH、甘油等醇类物质既是关键化工原料也是重要液体燃料,在可持续能源体系中扮演着重要角色。借助电化学手段,无须高温高压即可将醇类定向转化为对应的羧酸、醛或酮等具有更高附加值的化学品,并且可以使用风能、光能等可再生能源供电,推动绿色化工发展。与低碳烃相比,醇类分子的电氧化过程具有更高的催化效率,原因是醇类分子C—H和O—H活化更容易,电氧化所需要的过电位更低。
1.醇类分子电氧化的基本反应路径 完全氧化:醇类分子直接氧化为CO2 ,释放大量电子,常用于燃料电池发电过程,如直接甲醇燃料电池(Direct Methanol Fuel Cell,DMFC)。
部分氧化/升级反应:将醇类分子定向电氧化为醛、酮或羧酸,这类反应能够制备高附加值化学品,是近年研究的重点方向。以C2 H5 OH为例,它在电极表面可能经历以下转化:CH3 CH2 OH→CH3 CHO→CH3 COOH→CO2 ,定向控制的关键在于稳定中间体并避免过度氧化。多羟基醇(如甘油)的氧化路径更加复杂,可以生成多种含氧中间产物,如二羟基丙酮、甘油酸等,其产物选择性受电极材料、pH值、电位等因素显著影响。
2.CH3 OH的电氧化 Qin等[43] 通过一步水热法制备了Cr掺杂的α-Ni(OH)2 催化剂(Cr0.02 Ni(OH)2+ δ ),提出了一种通过调控局部配位结构以精准调节电化学界面微环境的新策略,实现了CH3 OH电氧化反应性能的显著提升,为CH3 OH向甲酸盐的高效定向转化提供了新的设计思路。不同于传统的纳米结构调控或表面功能化等微环境优化手段,该工作创新地引入硬路易斯酸Cr原子参与调控反应界面。一方面,Cr掺杂在催化剂表面构建了相对疏水的界面环境,可有效加速水分子解离动力学,并抑制孤立H2 O分子对活性位点的占据,从而避免反应位点被堵塞。另一方面,Cr与Ni之间的电子转移显著调节了Ni的d带中心位置,增强了催化剂对OH- 的吸附能力,可促进OH- 在带电界面处的富集,进而显著提高局部碱性环境。此外,Cr掺杂还可诱导活性相由α-Ni(OH)2 向结构无序的γ-NiOOH转变,增加OH– 吸附相关的活性位点数量,并有效降低CH3 OH分子解离的热力学能垒,从而优化反应的决速步,整体提升CH3 OH电氧化动力学。
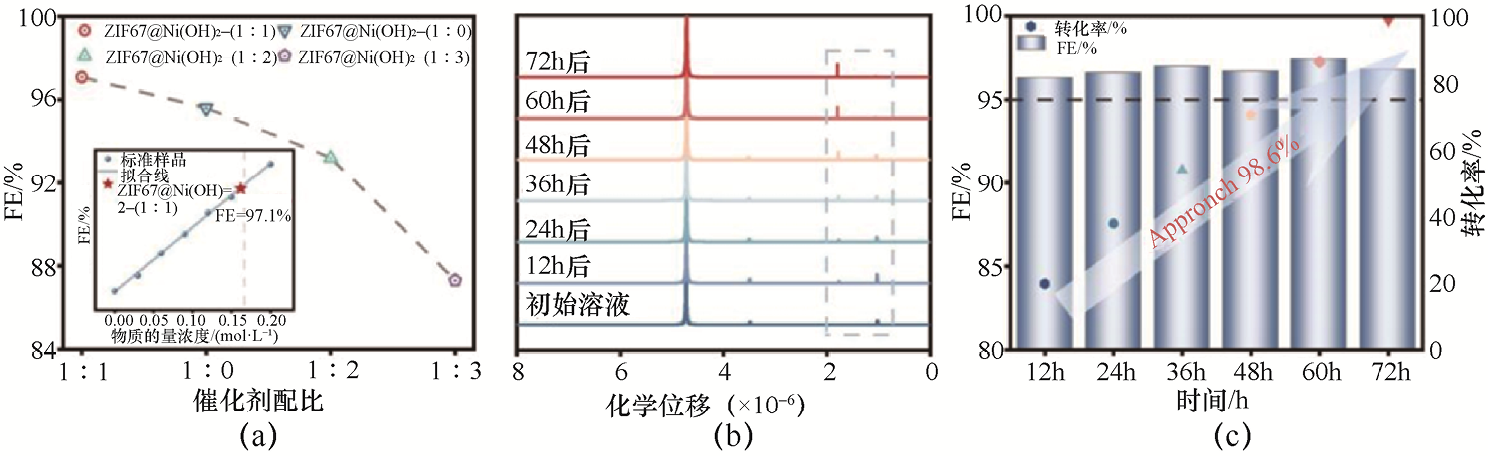
3.C2 H5 OH的电氧化 Zhu等[44] 通过提出一种混合溶剂热MOF保护策略,诱导了新型类晶格氧介导的C2 H5 OH电氧化反应路径。该工作利用混合溶剂调控体系极性的合成策略,创新地实现了在构建Ni/Co氢氧化物催化活性相的同时,有效保持MOF的结构优势,从而为MOF衍生电催化剂的结构设计与反应路径调控提供了新的思路。化学测试结果表明,ZIF67@Ni(OH)2 -(1∶1)在EOR中性能优异。1 H NMR与法拉第效率分析进一步证实,该催化剂在宽电位范围内可高选择性地将C2 H5 OH转化为CH3 COOH,法拉第效率超过97%,C2 H5 OH转化率接近99%(见图1-17)。这一结果表明,采用混合溶剂热MOF保护策略构建的Ni(OH)2 @MOF复合结构能够在保持高活性与稳定性的同时,实现高效、高选择性的C2 H5 OH电氧化。
图1-17 C2 H5 OH电氧化制备CH3 COOH[44]
(a)不同样品的C2 H5 OH电氧化法拉第效率,插图为ZIF67@Ni(OH)=2-(1∶1)的法拉第效率分析;(b)ZIF67@Ni(OH)2 -(1∶1)电极在不同选定时间点C2 H5 OH电氧化所得产物的1 H NMR谱图;(c)ZIF67@Ni(OH)2 -(1∶1)在不同时间点的法拉第效率与转化效率
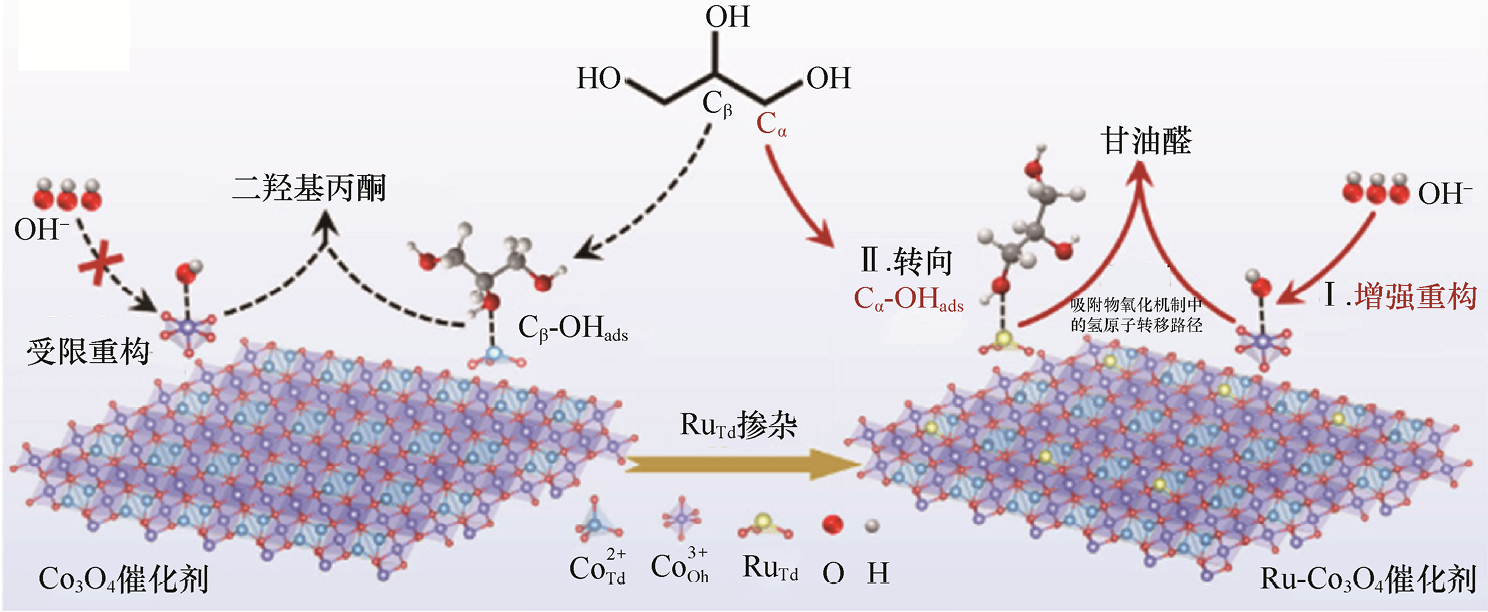
4.甘油及多羟基醇的电氧化 甘油是生物柴油生产的副产物,甘油电氧化可产生高价值化学品如甘油醛、甘油酸、乙醛酸等,被认为是生物质电化学转化的关键路径。Luo等[45] 将单原子Ru[0.36%(wt)]掺杂入Co3 O4 自旋结构的四面体位点,激活了钴氧化物晶格并显著提升了甘油电氧化性能。在这一策略中,通过Ru掺入Co3 O4 晶格,可显著增强催化剂表面在反应过程中的结构重构能力,原位生成大量Co3+ -(OH)ads 电亲和氧物种。这些活性氧物种不仅加速了甘油的吸附与初步氧化反应,还通过调控甘油的吸附构型,使甘油更易沿Cα -OH原位氧化路径发生选择性氧化,优先向甘油醛方向转化(见图1-18)。该策略中,Ru原子促进Co3 O4 晶格快速重构,形成富含Co3+ -(OH)ads 的界面活性位点网络,使催化剂在中性介质下对甘油电氧化表现出加速的动力学及优异的产物选择性。实验结果显示,Ru-Co3 O4 /NF催化剂在约-1.16V(相对于RHE)电位下实现了10mA·cm-2 的电流密度,并在1.3~1.7V(相对于RHE)保持了57.84%±2.12%的甘油醛产物选择性,同时总C3 产物选择性达90%以上。以上机理与性能数据表明,通过Ru掺杂激活Co3 O4 晶格并生成优先促进Cα -OH氧化的表面氧物种,可以显著增强甘油向甘油醛的电化学转化性能。
图1-18 Ru原子调控Cα -OH吸附构型并增强OH- 吸附富集的示意图[45]
1.3.4 醛类分子的电氧化 在化工生产与环境治理领域,HCHO、CH3 CHO等醛类化合物扮演着关键角色。通过电氧化技术,这些醛类分子不仅能够转化为羧酸等高价值产品,还可作为一种绿色环保的污染物处理手段。与醇类分子相比,醛类分子在氧化反应中展现出更低的反应过电位,意味着其更易被氧化。同时,该过程的产物分布也更具可控性,为化工合成与环境修复提供了高效且精准的技术路径。
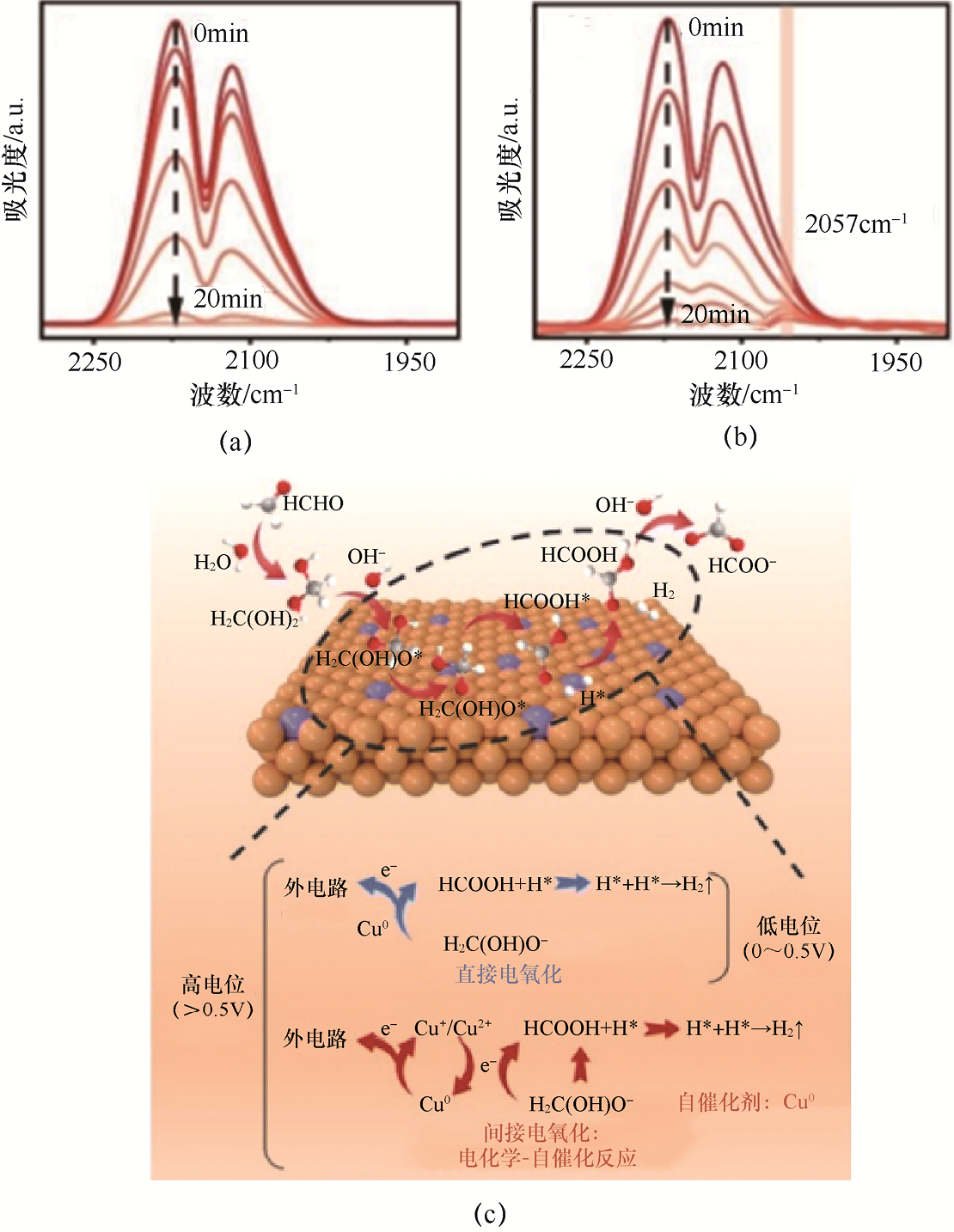
1.HCHO的电氧化 HCHO的电氧化过程中主要经过中间体HCOOH,并可进一步深度氧化至CO2 。在碱性环境中,OH– 的参与显著提升了反应速率,使得HCHO氧化反应(Formaldehyde Oxidation Reaction,FOR)能够在较低电位下高效进行。基于这一特性,将FOR与HER耦合,形成阴阳极协同产氢的策略,展现出良好的应用前景。然而,该体系在实际运行中面临催化剂稳定性问题:在高电位下,Cu0 迅速被电氧化为Cu+ /Cu2+ ,但其自发还原为Cu0 的过程缓慢,导致活性Cu0 位点逐渐减少,催化惰性的Cu+ /Cu2+ 物种不断积累,最终引起催化剂失活。这一动力学失衡显著压缩了FOR的有效电位窗口,限制了其在较高电流密度下的持续运行。为解决该问题,Song等[46] 设计了一种在铜泡沫上负载高度分散Pt位点的催化剂(Pt1 /Cu-CF)。该催化剂能够保留足够的Cu0 活性位点,同时利用Pt优异的C—H断裂能力及高理论氧化电位,增强整体催化性能。在CO脱附原位红外光谱测试[见图1-19(a)(b)]中,Pt1 /Cu-CF催化剂表面同时存在Cu0 和Cu+ 特征吸附峰:在2070cm-1 处观察到显著增强的Cu0 线性吸附峰(较纯Cu-CF提高约30%),表明Pt单原子位点的引入有效稳定了金属铜活性位点;与此同时,2100cm-1 处仍可检测到微弱的Cu+ 桥式吸附信号,暗示在电氧化还原过程中形成了动态的Cu0 -Cu+ 自催化循环。这种双价态共存现象与Pt的电子耦合效应密切相关,Pt的强吸电子特性既促进了C—H活化能力,又通过局域电荷转移抑制了Cu0 的过度氧化,最终实现了高活性位点密度与自再生能力的协同优化。基于此,该研究提出了Cu基催化剂在广泛电位窗口内的HCHO氧化机理(见图1-19):在低电位条件下,Cu位点主要通过直接氧化HCHO生成产物;而在高电位条件下,反应包含直接与间接氧化过程,Cu在Cu0 、Cu+ 和Cu2+ 之间动态变化,并通过自催化路径部分还原为Cu0 ,从而维持活性位点的持续供给与高效催化。该研究揭示了Pt修饰Cu催化剂的协同作用机理,为高电位下HCHO氧化提供了设计参考。
图1-19 CO脱附过程中原位红外光谱测试和HCHO电氧化反应机理分析[46]
(a)Cu-CF上的原位红外光谱图;(b)Pt1 /Cu-CF上的原位红外光谱图;(c)Cu基材料在宽电位范围内用于阳极产氢的HCHO电氧化反应机理示意图
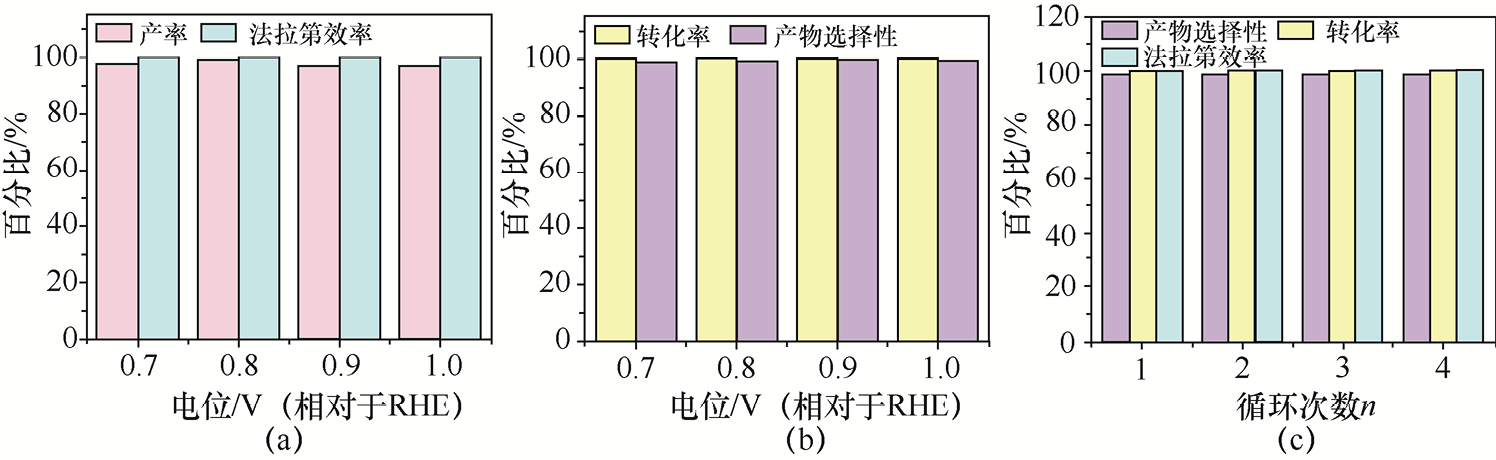
2.其他醛类物质的电氧化 Yao等[47] 采用水热反应和可控温度煅烧策略,在泡沫镍(Foamed Nickel,常用NF表示)载体上构建了Ag纳米粒子-NiO纳米线异质结催化剂(Ag-NiO/NF)。电化学测试结果显示,Ag-NiO/NF对5-羟甲基糠醛(5-Hydroxymethyl Furfural,HMF)、糠醛、HCHO、CH3 CHO及苯甲醛等多种醛类分子呈现优异的电氧化活性,而对相应醇类分子没有任何电化学响应,证实了其高选择性催化特性。更重要的是,在HMF氧化反应(HMF Oxidation Reaction,HMFOR)中,Ag-NiO/NF的电流密度达到10mA·cm-2 所需电位仅为0.45V(相对于RHE),获得的目标羧酸类产物的产率为99%,产物选择性和法拉第效率均达到100%左右(见图1-20)。原位红外光谱、电化学阻抗谱及理论计算共同揭露了Ag与NiO形成的异质界面能有效促进HMF分子与OH⁻在催化剂表面共吸附和关键中间体生成,最终实现优异的催化活性。
图1-20 Ag-NiO/NF催化剂的HMFOR性能研究[47]
(a)Ag-NiO/NF催化剂不同电位下HMFOR的产率和法拉第效率;(b)Ag-NiO/NF催化剂不同电位下HMF的产物选择性和转化率;(c)Ag-NiO/NF催化剂的HMFOR稳定性测试
3.机理研究与理论模拟 理论计算研究表明,醛类分子的电氧化通常经历分子在催化剂表面的吸附与后续羟基化等关键步骤,而这些步骤往往决定了整个反应的决速步。以HCHO在NiOOH电极上的转化为例,原位光谱及DFT计算均表明,HCHO首先在电极表面吸附并生成中间体*HCOO,随后可进一步氧化为HCOOH或完全氧化为CO2 [48-49] 。相比之下,CH3 CHO的电氧化路径呈现出不同的能垒控制特征:DFT计算表明CH3 CHO→CH3 COO- 的氧化步骤具有较低的反应能垒,使得CH3 COOH(或CH3 COO- )成为主要产物[50] 。进一步的理论模拟与原位谱学联合分析强调,电极表面的结构特征(包括缺陷位点、配位不饱和中心及晶格应力等)能够显著影响反应中间体的吸附强度与稳定性,从而调节关键步骤的反应能垒,并最终决定产物选择性[51-52] 。这些发现为理性设计高效且高产物选择性的电催化体系提供了可靠的理论依据。
1.4 电解槽设计电解槽是连接催化材料与反应过程的核心载体,它的结构设计直接决定传质效率、离子传输速率与产物分离效果,最终影响反应的法拉第效率、电流密度、能耗与稳定性等关键指标。针对电化学碳循环反应中气体反应物/产物共存、多相界面反应主导的特点,目前主流电解槽可分为H形电解槽、流动电解槽与MEA三类[53] 。三者在结构复杂度、性能参数与应用场景上呈现清晰的技术演进逻辑,分别对应基础研究、中试开发与工业化应用这3个核心阶段。
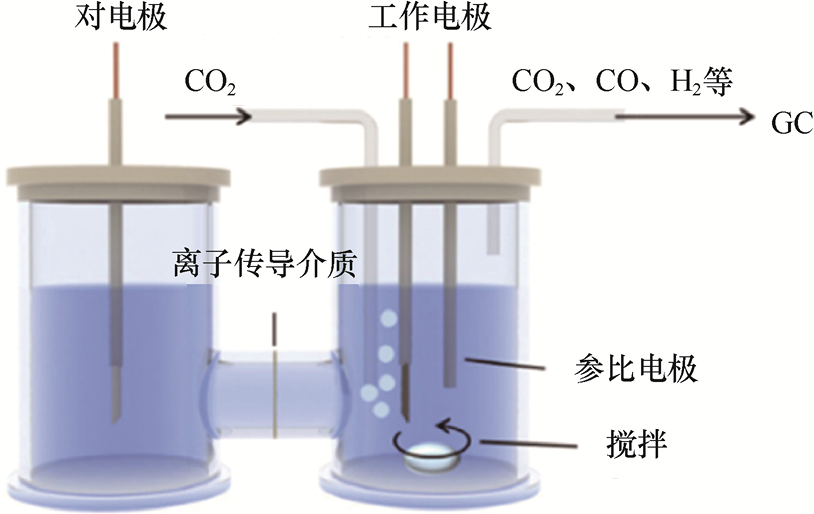
1.4.1 H形电解槽 H形电解槽因经典的H形腔室结构得名,是电化学研究中应用最广泛的实验室级反应器。它的核心设计是通过离子传导介质[质子交换膜(Proton Exchange Membrane,PEM)或盐桥]将两个独立腔室(阴极室、阳极室)连接,阴极室与阳极室分别容纳工作电极(如CO2 RR催化剂)与对电极(如Pt片、石墨棒),参比电极直接插入阴极室以精准监测电位,形成完整的三电极测试体系(见图1-21)[53] 。
图1-21 H形电解槽的结构[53]
1.结构设计与核心特性 H形电解槽的优势主要体现在腔室隔离机制、电位测量精度,以及材料选择与结构变体3个方面。
(1)腔室隔离机制。阴极与阳极通常通过全氟磺酸树脂膜(常被称为Nafion膜)、玻璃砂芯或盐桥相连,实现离子传输而不发生溶液混合,从而显著降低产物交叉扩散的风险。在CO2 RR体系中,这类结构可将阴极生成的还原性产物(如CO、C2 H4 )与阳极析出的O2 完全分开,令后续产物检测与定量更加可靠。
(2)电位测量精度。参比电极(如Ag/AgCl或Hg/HgO)可布置在靠近工作电极的位置,减弱由溶液电阻引起的电位偏移,使测得电位的误差通常保持在±5mV以内,为过电位分析与催化剂性能评估提供较高的准确性。
(3)材料选择与结构变体。H形电解槽多采用耐腐蚀性良好的硼硅玻璃,便于观察反应进程。根据研究需求的不同,H形电解槽的结构也可作调整。例如,简化的单室结构能够减小溶液电阻,用于某些高导电体系;定制化版本则可预留光学窗口,方便耦合原位拉曼等表征手段。
2.性能局限的根源 H形电解槽在电化学碳转化应用中的局限主要体现在传质能力不足与难以实现放大两个方面,根源在于该装置偏向静态体系,而CO2 RR需要更高效的物质输运与更接近工业工况的环境。
(1)传质受限。在水溶液中,CO2 的溶解度仅数十毫摩尔级(约33mmol,25℃、1atm),并依赖自然扩散抵达电极界面,扩散层厚度往往达到数百微米。这种缓慢的物质输运使阴极附近的CO2 浓度长期偏低,导致可实现的电流密度普遍受限(约50mA·cm-2 ),明显无法满足产业化对高产率的要求。
(2)长期运行受液相环境扰动。反应过程中,阴极侧CO2 的持续消耗会引发局部碱化(如生成
(3)难以跨越尺度的性能外推。H形电解槽多数采用0.1~1cm2 的小面积电极,边界效应对传质及电流分布影响明显,使实验条件与实际工业反应器相差较大。因此,从该体系获得的数据通常无法直接用于预测大规模电解系统的表现。
3.不可替代的应用场景 尽管性能受限,H形电解槽仍是实验室基础研究中不可替代的工具,核心价值在于低成本与高可控性的平衡。
(1)催化剂初步筛选:可快速对比不同催化剂的活性(过电位)与产物选择性(法拉第效率)。例如,在Cu基CO2 RR催化剂改性研究中,通过H形电解槽可在1~2天内完成10余种催化剂的初步排序。
(2)反应机理探索:适配同位素标记、原位电化学红外等机理研究手段。例如,利用13 CO2 作为反应物,在H形电解槽中追踪碳源流向,明确*COOH、*CH3 等中间体的生成路径。
(3)反应条件优化:可系统探究电解质种类(KCl、KHCO3 )、物质的量浓度(0.1~1mol·L-1 )、温度(25~80℃)等参数的影响规律,为后续高等级反应器提供基础参数支撑。
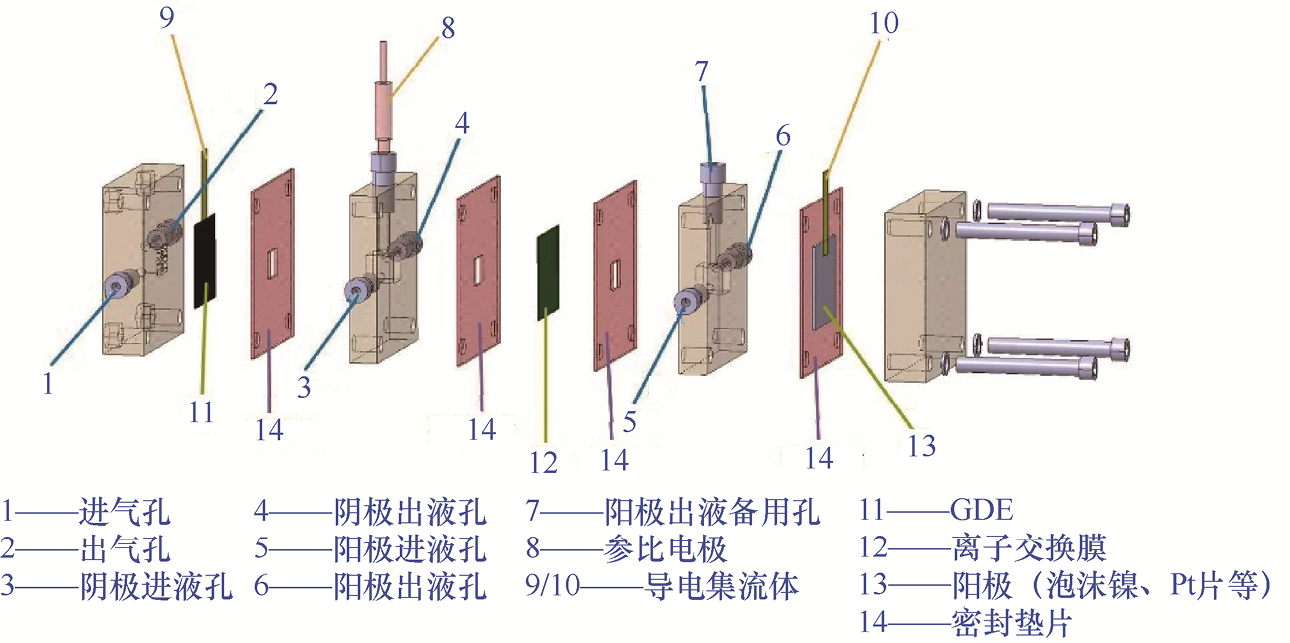
1.4.2 流动电解槽 流动电解槽基于“主动传质-三相界面”的概念设计,突破了H形电解槽的传质限制,成为连接实验室基础研究与工业化应用的核心中试平台。该电解槽的核心创新点是将工作电极升级为气体扩散电极(Gas Diffusion Electrode,GDE),并通过泵驱动电解液强制流动,构建高效的气-液-固三相反应环境。流动电解槽的结构如图1-22所示。
图1-22 流动电解槽的结构
1.结构设计与传质强化机制 流动电解槽的性能优势源自基于“组件协同-流场调控”的系统设计。它的核心结构通常包含阴极气体腔室、阴极电解液腔室与阳极腔室3个部分,通过GDE与离子交换膜实现腔室隔离。GDE由气体扩散层(Gas Diffusion Layer,GDL)和催化剂层(Catalyst Layer,CL)构成。GDL采用碳纸/碳布,负责CO2 传输与电子传导;CL是被均匀喷涂或刮涂于GDL表面的催化剂浆料,能够被电解液充分浸润。CO2 直接通过GDL的多孔结构到达CL表面,无须经历溶液扩散过程,可极大地提升传质效率。同时,电解液以5~50mL·min-1 的流速在CL表面流动,及时补充消耗的反应物并脱附产物(如避免*CO在Pt表面堆积),可使电流密度达300~1000mA·cm-2 ,较H形电解槽提升10倍以上。此外,流道板的设计直接影响传质均匀性,目前主要采用3类流道:蛇形流道(压力损失大,但传质均匀性好)、平行流道(压力损失小,适用于大面积电极)、仿生流道(模拟叶脉结构,兼顾均匀性与低阻力)。
2.关键组件的设计准则 流动电解槽的整体性能高度依赖各关键组件之间的协同匹配,各部件需同时满足高效传质、稳定导电与长期耐腐蚀等多重要求。GDL是实现气体反应物高效供给的核心组件,通常需要兼具良好的透气性与导电性,同时具备一定的疏水性,以防止电解液渗入并阻塞气体传输通道。实际应用中,常采用经疏水改性的碳纸或碳布作为GDL材料,通过调控其孔结构和表面润湿性,实现气-液界面的稳定构建。
CL通常是“催化剂-导电碳载体-离聚物”结构的复合体系。其中,离聚物主要承担离子传导和结构粘接的作用,需要精细调控含量。离聚物含量过低会限制离子传输效率,含量过高则可能堵塞多孔结构,削弱气体扩散与活性位点的可达性,从而影响整体反应动力学。
流道板在流动电解槽中承担电流收集和反应物分配的双重功能,材料选择需在导电性与耐腐蚀性之间取得平衡。人们常通过优化流道的形状、宽度与深度,使电解液和气体在反应区域内均匀分布,避免局部滞流或产物积累,以提升电化学反应的稳定性与连续运行能力。
3.性能优势与应用边界 流动电解槽的优势在于能够在实验室尺度下构建更接近工业条件的反应环境,同时保留对操作参数的灵活调控能力。在用于CO2 RR生成多碳产物时,流动体系通常能够显著增强气–液–固三相界面的传质,与传统H形电解槽相比更容易维持高反应速率和较高的多碳产物选择性。在优化的界面结构与流体条件下,系统可以在较高的电流密度下稳定运行,并展现出比较理想的C2+ 产物法拉第效率,从而突破H形电解槽受CO2 供应限制而难以实现高产率的瓶颈。在电氧化反应(如AOR)中,电解液的循环流动保证了底物的持续输运,通常能够支持较大的工作电流密度,并在长时间运行过程中维持比较稳定的产物输出。得益于流体化的供料方式和可控的停留时间,流动电解槽在放大后催化剂性能评估(电极面积可扩展至数平方厘米)以及多参数联合优化(如流速、温度与压力的耦合调节)方面具有显著优势。同时,该电解槽便于与在线气体采集或初级分离单元集成,为探索产物分离与利用策略提供了便利。
尽管整体性能优越,流动电解槽仍面临一些工程性限制。例如,该电解槽通常需要一定体积的循环电解液才能保持稳定运行,装置相对庞大;此外,若流场设计不当,局部区域可能出现干燥或水淹等现象,从而导致反应界面不均一,并影响催化剂稳定性。因此,在设计与使用该电解槽时,需要在传质、流体分布和界面湿润性之间实现合理平衡。
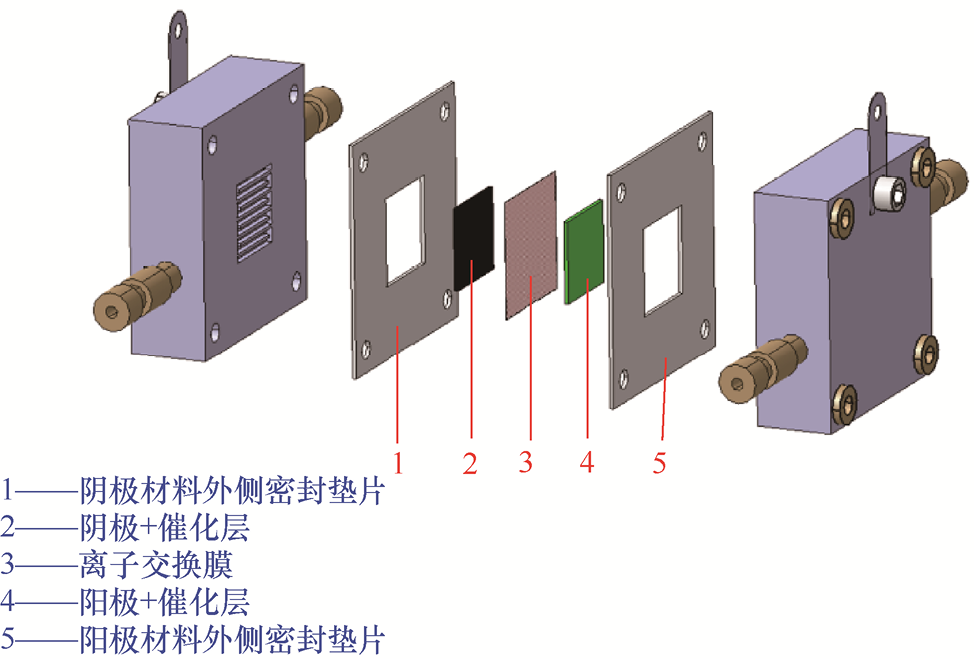
1.4.3 MEA电解槽 MEA电解槽以MEA为核心功能部件,通过“催化剂层-膜-催化剂层”的三明治结构,将电极、电解质与离子传导介质(离子交换膜)融为一体,彻底摒弃独立电解液腔室,是电化学碳循环工业化的核心技术载体。MEA的结构如图1-23所示。
图1-23 MEA的结构
1.结构设计与性能提升原理 MEA的优势主要源自其在结构集成、传质距离和能效管理方面的协同提升。它的核心结构特征:阴极CL(通常加载于GDL)与阳极CL通过热压或直接涂覆的方式复合在离子交换膜的两侧,使得整体反应单元高度集成,厚度可控制在极薄的范围内,与传统的分室式电解槽相比显著缩小了体积,提高了系统的紧凑性与反应器利用率。
在离子传输方面,MEA将阴极催化层与阳极CL紧密贴合,使得质子或阴离子的传输距离从毫米级缩短至微米级。较短的传输路径通常能够有效降低欧姆电阻,有助于在相同电流密度下实现更低的槽压(单个电解槽中阳极和阴极之间的电压差)与更高的能量利用率。因此,在MEA电解槽中,整体电能消耗往往比传统流动电解槽更容易优化。
产物管理也是MEA电解槽的一项优势。由于反应主要在GDL界面进行,阴极生成的气态产物可以直接穿过多孔结构迅速逸出,通常不需要经过大量电解液的溶解或萃取过程;液态产物则随局部生成的水一并排出,分离过程相对简单。这种设计降低了对大体积电解液的依赖,使得后处理步骤更适合与连续生产工艺对接。
2.工业化进程中的核心挑战 MEA电解槽的产业化仍面临界面一致性、水管理以及整体成本等方面的限制。首先,在CL制备过程中,如果离子交换膜与CL之间的接触不够紧密,容易形成微尺度空隙,使得气体在界面处发生不必要的混合或滞留,进而影响传质效率。此外,界面离子通道的不连续性会增加传导阻力,使整体电解性能难以充分发挥。
水管理也是MEA电解槽运行的关键挑战之一。反应过程中生成的水需要通过GDL有效排出,以保持气体通道的通畅;与此同时,膜材料又必须维持适度湿润,以保证离子传输的稳定性。如果水分排出过快,会导致膜局部干燥,使电阻升高并影响反应电位控制;排出不足则可能出现水分积聚,使CL通道被堵塞,从而抑制反应物的传递。虽然通过调节扩散层的疏水性、平衡湿气供应或采用脉冲操作可以缓解这些问题,但在高电流密度条件下,水的产生与移除仍然难以保持长期平衡。
在成本构成方面,离子交换膜与催化层通常是电化学系统中费用占比较高的核心部件,尤其是PEM以及部分采用贵金属或复杂制备工艺的催化剂体系。随着装置运行时间的延长,催化层可能发生结构重排或活性位点流失,离子交换膜则可能因化学与机械应力而逐步老化,同时反应界面局部环境的波动也会引发性能衰减。这些因素共同限制了体系的长期稳定运行,使其在连续或大规模应用中面临可靠性挑战。
当MEA尺寸进一步放大时,MEA电解槽内部的传质与电流分布更难保持均匀,局部区域可能出现显著的反应速率与电流密度差异,从而诱发温度和电化学状态不均匀。这种不均匀性不仅会降低整体能效,还可能加速离子交换膜与CL的老化,成为制约系统规模化与长期运行的重要工程瓶颈。针对这一问题,章俊良团队[54] 提出了适用于大尺寸MEA的流场设计原则,包括提升CO2 分布均匀性、增强垂直方向(法向)传质能力以及减少积水现象,为规模化电堆设计提供了重要参考方向。
3.工业化应用与技术展望 MEA凭借结构高度集成、传质距离短和能效优势,在实验室与工程放大研究中都展现出向工业化过渡的潜力。它具有模块化、紧凑化的特征,适用于多种规模化场景。例如,在面向大型碳转化装置的概念设计中,通常会采用由多片MEA组成的电解堆,以实现较高的CO2 处理通量和能量利用效率;在空间、海洋等受体积和能耗限制的应用环境中,紧凑型MEA装置能够在有限体积内提供较高的能量密度和较强的反应能力,因此被视为潜在的系统集成方案。在工程规划中,MEA的未来技术路线常以“高电流密度+长寿命+低成本”三重目标为核心,例如追求安培级电流密度、万小时量级的运行寿命,以及更低的单位功率催化剂成本。这些指标并非当前技术水平的直接反映,而是行业在长期发展中希望逐步实现的目标,用于指导材料设计、系统结构优化与产业化策略制定。
1.4.4 横向对比与技术演进 H形电解槽、流动电解槽与MEA电解槽在性能特征与应用定位上呈现出清晰的梯度关系,共同构成电化学碳循环技术从实验室研究到工程化应用的反应器体系。这3类电解槽在设计理念和功能侧重点上的差异,主要体现在传质方式、结构集成度以及可放大性等方面,演进逻辑可概括如下。
(1)H形电解槽以结构简单、操作灵活为特点,适用于精确控制电位和反应条件,便于解析反应机理和评估催化剂本征活性。但由于气液传质主要依赖溶解扩散,电流密度和体积功率密度较低,难以满足高负载运行和规模化需求,通常局限于短时实验和基础研究阶段。
(2)流动电解槽通过引入GDE和连续流动电解液,显著强化了CO2 供给和产物移除过程,能够以更快的反应速率稳定运行,适合开展高电流密度条件下的性能测试与工艺参数优化。然而,该电解槽通常依赖体积较大的液相电解质,系统复杂度和运行稳定性仍有待进一步提升。
(3)MEA电解槽代表了高度集成化的发展方向,将CL、离子传导与气体输运紧密耦合,能够在较低欧姆损失和较高能效下实现连续运行,具备良好的模块化与放大潜力。与此同时,它对界面结构、水管理与材料稳定性的要求较高,制造成本和运行控制难度也相应较高。
总体而言,电解槽技术的发展路径体现为:从H形电解槽侧重机理认知与材料筛选,到流动电解槽强调性能验证与工况优化,再到MEA电解槽聚焦系统集成与工程化应用。这一发展过程始终围绕传质强化、结构集成与成本控制三大核心目标展开,推动电化学碳循环技术逐步从实验室研究迈向实际应用场景。
1.5 挑战与展望电化学碳循环技术依托外加电能实现含碳小分子的定向转化,构建起“捕集-转化-再利用”的闭合流程,被视为缓解化石能源依赖与降低碳排放的重要路径。尽管近年来,人们在材料设计、反应机理与产物选择性方面取得了大量实验室突破,但距离真正的工程化应用仍存在材料、器件以及系统集成等多层面的差距。系统梳理当前核心挑战并明确未来发展方向,对推动该技术从科研探索迈向规模化应用具有重要战略意义。
1.5.1 核心挑战 尽管电化学碳循环技术在催化材料开发、反应器设计等领域已取得阶段性成果,但面向工业化需求,催化剂、反应器与系统集成三大核心层面仍存在亟待突破的瓶颈,这些挑战相互交织,共同制约着技术的规模化应用。
1.催化剂层面:活性、产物选择性与稳定性的“三角困境” 作为电催化反应的核心驱动力,催化剂的性能优化始终面临活性与产物选择性的权衡、稳定性衰减的多因素耦合、成本与活性的相互制约这三大挑战。
(1)活性与产物选择性的权衡。电催化反应的多电子转移特性导致产物分布复杂,催化剂难以精准调控反应路径。例如,Cu基催化剂虽能实现CO2 至高价值C2+ 产物(C2 H4 、C2 H5 OH)的定向转化,但其过电位通常超过0.4V,且伴随CH4 、CO等副产物生成,法拉第效率难以突破90%;对CO2 至C1 产物的定向转化具有高活性的Au基催化剂,又因电子转移路径受限,无法高效转化为长碳链产物。这种权衡本质上源自催化剂表面中间体吸附能的“火山形曲线”关系。对关键中间体(如*COOH、*CO)的吸附强度过高或过低,均会导致反应活性或产物选择性下降[8] 。
(2)稳定性衰减的多因素耦合。在复杂的电化学环境中,催化剂通常会受到多种降解机制的共同影响,从而导致性能随时间下降。贵金属催化剂可能因电化学腐蚀或表面重构而逐渐流失活性位点,而非贵金属纳米催化剂易因表面能驱动而发生团聚,使比表面积和活性位点密度降低。此外,反应中间体的强吸附、电解液成分引发的沉积或侵蚀,以及外界操作条件变化,都可能导致催化剂中毒、结构被破坏或载体界面退化。这些因素通常会叠加作用,使催化体系在连续运行中出现显著的活性和产物选择性下降,运行寿命受限。
(3)成本与活性的相互制约。高性能电催化体系往往依赖贵金属元素来实现高活性和高稳定性,但其价格昂贵、资源稀缺,使催化剂成本在系统构成中占据很大比例。相比之下,非贵金属体系具备原料廉价、可规模化制备的优势,但通常在活性、稳定性和长时间运行能力方面与贵金属体系存在差距,且在严格工况下更容易失活。这种“高性能材料成本高、低成本材料性能不足”的矛盾,使催化剂设计成为限制工业化推进的主要瓶颈之一。
2.反应器层面:传质、调控与放大的“工程瓶颈” 作为连接催化剂与反应体系的核心载体,反应器的设计必须同时解决多相传质、操作条件调控及规模化放大的工程挑战,但现阶段技术仍面临明显不足。
(1)高电流密度下的传质极限。在流动电解槽与MEA电解槽等反应器中,随着工作电流密度不断提高,多相传质逐渐成为性能瓶颈。气体在GDE内部的输运路径受微结构影响而不均一,容易出现反应物供应不足的区域。与此同时,产物(如C2 H4 、H2 O)在CL中滞留,会降低活性位点的可利用性并促使副反应发生。对复杂多电子转移过程(如CO2 RR)而言,局部产物堆积不仅抑制目标产物生成,还可能改变产物选择性,使整体性能难以在高负荷下保持稳定。
(2)水-热-电的协同调控难题。电化学反应中,质子传输、水生成/迁移与焦耳热释放形成复杂的耦合效应,难以精准调控。在碱性CO2 RR中,阴极生成的OH- 与CO2 反应生成HCO3 - /CO3 2- ,既会降低反应物利用率,又易在流道内形成盐沉积,使孔隙堵塞。MEA电解槽中,反应生成的水若无法及时排出,会导致“水淹”CL;缺水则会使离子交换膜变干,导致电阻骤升。这两种情况均会使MEA电解槽的槽压升高,能效下降。同时,局部热量堆积引起的温度梯度会影响反应动力学和产物选择性,使体系响应更加复杂。
(3)规模化放大时的性能衰减。实验室级小面积(1cm2 以下)电极的测试结果往往难以平移至大面积(1m2 以上)工业组件。随着尺寸变大,流场分布趋于不均,局部区域可能承担异常高负荷而出现热点效应,导致部件老化加剧。此外,随着电极与离子交换膜接触面积的增加,界面电阻增大现象会更加显著,使放大后的装置性能明显低于实验室单元。这种规模化放大导致的性能衰减,是阻碍技术落地的主要工程瓶颈之一。
3.系统集成层面:能效、分离与耦合的“全链制约” 电化学碳循环是一种涵盖“捕集-电解-分离-利用”的完整技术链条,但当前各环节之间的协同不足,导致整体能效偏低、运行成本偏高。
系统能效在链条间逐级损耗。从CO2 捕集到电化学转化再到产物分离,每个环节都存在能源输入与效率损失。电解槽本身的电化学极化、欧姆损耗和副反应会消耗大量电能;CO2 捕集与提纯需要额外的热能或电能输入,使前端处理的成本与能耗升高;多组分产物的分离和净化又需要复杂的分离技术,使后端能耗进一步扩大。这种多环节累积效应使系统总能效明显低于单一工序能效,成为制约技术推广的主要障碍。
产物分离的高成本壁垒。CO2 RR等反应常生成多组分产物(如CO、CH4 、C2 H4 、HCOOH),不同产物的物理化学性质相近,分离难度极大。复杂的气体混合物通常需要多级分离过程,而该过程对设备投资、能源输入和运行维护均提出较高要求;液态产物的浓缩涉及大量能耗或复杂工艺,使分离成本始终保持高位。因此,在许多体系中,产品分离与提纯的投入甚至可能超过电解过程本身,成为决定经济性的关键因素。
与可再生能源的适配鸿沟。太阳能和风能的输出具有间歇性和波动性,而电化学反应对稳定的电流密度与电位的依赖度极高。当驱动电源波动较大时,会引起反应体系内部电位变化,从而影响反应路径与产物分布,使效率、产物选择性和稳定性发生显著波动。虽然储能系统可以在一定条件下缓解这一矛盾,但其成本、体积与寿命往往难以满足大规模装置连续、稳定运行的需求,这使得可再生能源与电化学碳循环技术在工程层面面临适配难题。
1.5.2 发展方向 针对上述挑战,未来需通过催化剂创新突破性能瓶颈、通过反应器优化提高工程效率,推动电化学碳循环技术从实验室走向工业化。
1.催化剂创新:原子级调控与非贵金属替代 催化剂研发需从经验筛选转向精准设计,通过多尺度调控实现活性、产物选择性与稳定性的协同优化,同时降低成本。
(1)原子级活性位点工程。单原子与双原子催化剂因其明确的活性位点和高原子利用率,成为提升性能的关键方向。精细调控原子周围的配位环境,可以显著改善反应的活性与产物选择性。未来,结合理论计算和原位表征,实现原子级位点电子结构与配位环境的精准设计,有望解决活性与产物选择性之间的权衡问题。
(2)非贵金属催化剂的性能跃升。基于过渡金属的催化剂是降低成本的主要路径。调控纳米结构、载体相互作用及界面电子效应,可以显著增强活性和稳定性。例如,将金属纳米颗粒负载于载体上可抑制团聚,延长催化剂寿命;复合材料的电子传导增强效应,有助于提高催化活性。同时,新型电解质(如离子液体)或表面修饰策略可优化催化剂微环境,进一步改善性能。
(3)催化剂的多功能集成设计。面对复杂的反应环境,催化剂需要兼具催化活性、抗腐蚀与抗中毒功能。例如,在催化剂表面构建保护层,既可保证反应物传输,又能抑制材料溶解;引入辅助组分可清除表面吸附的中间体,提高耐中毒能力。未来,结合加速寿命测试和“结构-稳定性”模型,将有助于指导长寿命催化剂的设计。
2.反应器优化:传质强化与智能化调控 反应器设计需要突破传统工程思维,结合新型结构与智能技术,实现传质效率、参数调控和规模化放大的协同优化。
(1)传质强化的结构创新。开发三维多孔电极和仿生流道,可构建高效传质路径。凭借高比表面积和贯通孔隙结构,泡沫金属或多孔碳基电极可显著增加气-液-固三相界面面积;通过模拟植物输运系统设计的叶脉状仿生流道可提高反应物分布均匀性,从而提升传质效率。此外,新型GDL的疏水-亲水梯度设计,可有效抑制水堵塞和膜干问题,保证高电流密度条件下的稳定运行。
(2)智能化监测与动态调控。集成原位表征技术与人工智能算法,可实现反应过程的精准调控。例如,在反应器中嵌入原位拉曼光谱与XPS传感器,可实时监测催化剂结构演变与中间体生成;基于多参数数据集3版(Multi-Parameter Dataset Version 3,简称MED3)训练AI模型,可预测不同操作条件下的反应性能,动态调整电压、流速、湿度等参数。
(3)模块化与标准化放大。建立反应器设计标准体系,开发可拼接的模块化装置,以降低规模化难度。通过标准化单模块设计,可灵活调整产能,同时保持模块间流道的一致性,从而减少放大过程中的性能衰减。未来,可结合数值模拟和中试实验,建立“小试-中试-工业化”的放大准则,明确流场、电极尺寸等关键参数的缩放规律。
(4)系统集成与全链能效优化。系统设计应打破环节割裂,通过全链条协同和场景化定制,实现整体能效提升、成本降低和多元应用拓展。
(5)低能耗碳捕集与电解耦合。将电化学碳捕集与电解转化直接耦合,大幅降低系统能耗。谢和平院士团队[55] 开发的解耦电化学碳捕集技术,通过电化学-化学双步反应策略,避免氧气干扰,将捕集能耗降至每吨CO2 1.12GJ,且可直接输出高纯度CO2 供电解使用。这种“捕集-电解”一体化设计,可提升全链条能效,显著降低运行成本。
(6)产物高值化与分离简化。开发“一步法”转化技术与耦合工艺,跳过中间产物分离步骤。中国科学院团队[56] 开发的“电催化+生物催化”人工海洋碳循环系统,可直接将海水中的CO2 转化为可降解塑料单体(琥珀酸、乳酸),无须复杂分离即可合成聚丁二酸丁二醇酯、聚乳酸等材料,大幅降低流程成本。
(7)可再生能源的智能耦合。构建“发电-储能-电解”协同系统,应对可再生能源的间歇性。利用AI调度算法匹配光伏/风电输出与电解需求,光照充足时优先通过CO2 RR制备燃料,电能盈余时存储于电池,电能不足时通过燃料发电补充,形成“电-化学-电”闭环。同时,开发宽电位窗口催化剂与自适应电解槽,可耐受不同程度的电压波动,降低对储能系统的依赖。
(8)多元场景的定制化开发。针对不同场景需求,开发专用系统。例如,在深空探索领域,基于MEA的小型化系统可利用火星大气中的CO2 生成甲烷燃料与氧气,支撑地外生存;在化工园区,与工业尾气处理系统耦合,实现“以废治废”的碳转化;在海洋领域,利用海水作为碳源与电解液,开发可移动的碳转化平台,拓展应用边界。
电化学碳循环技术利用电能实现碳的定向转化,构建了“减排-能源-材料”一体化体系,是推动能源转型的重要技术支撑。尽管该技术仍面临催化剂性能优化难题、反应器工程化瓶颈及全链条系统制约,但材料科学、化学工程与人工智能的交叉应用为解决这些问题提供了新思路。
未来10~15年,随着原子级催化剂的精准设计、智能化反应器的实际应用以及全链条系统的优化,电化学碳循环技术有望实现从实验室验证到工业化应用的跨越。催化剂成本有望显著下降,稳定性可达到长期运行水平;反应器的电流密度和能效将大幅提升;全链条生产成本将具备商业可行性。最终,该技术有望形成以电化学为核心的绿色化工体系,不仅支撑传统产业的脱碳改造,还将为新能源、新材料以及外太空资源利用等领域提供发展空间,推动社会迈向可持续的碳平衡未来。
参考文献 [1] International Energy Agency. CO2 Emissions in 2022 (IEA) [EB/OL]. (2023-03-02) [2025-12-23].
[2] FANG W, LU R, LI F, et al. Low-coordination nanocrystalline Copper-based Catalysts through theory-guided electrochemical restructuring for selective CO2 reduction to ethylene[J]. Angewandte Chemie International Edition, 2024, 136(16): e202319936.
[3] DE LUNA P, HAHN C, HIGGINS D, et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes[J]. Science, 2019, 364(6438): eaav3506.
[4] SEH Z W, KIBSGAARD J, DICKENS C F, et al. Combining theory and experiment in electrocatalysis: insights into materials design[J]. Science, 2017, 355(6321): eaad4998.
[5] WANG H, MA C, LU Q, et al. Precise tuning of functional group spatial distribution on porphyrin rings for enhanced CO2 electroreduction selectivity[J]. Angewandte Chemie International Edition, 2025, 64(19): e202501091.
[6] NITOPI S, BERTHEUSSEN E, SCOTT S B, et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte[J]. Chemical Reviews, 2019, 119(12): 7610-7672.
[7] PETERSON A A, NØRSKOV J K. Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts[J]. The Journal of Physical Chemistry Letters, 2012, 3(2): 251-258.
[8] ZHANG R, KONG X, REN R, et al. Recent advances in copper-based catalysts for electrochemical carbon dioxide reduction to C2+ products[J]. Carbon Neutralization, 2025, 4(5): e70041.
[9] SIKDAR N. Electrochemical CO2 reduction reaction: comprehensive strategic approaches to catalyst design for selective liquid products formation[J]. Chemistry-A European Journal, 2024, 30(66): e202402477.
[10] CHATTERJEE S, GRIEGO C, HART J L, et al. Free standing nanoporous palladium alloys as CO poisoning tolerant electrocatalysts for the electrochemical reduction of CO2 to formate[J]. ACS Catalysis, 2019, 9: 5290-5301.
[11] HE F, CHEN X, XUE Y, et al. Theoretical prediction leads to synthesize GDY supported InOx 2 reduction[J]. Angewandte Chemie International Edition, 2024, 63(21): e202318080.
[12] LIU T, LUO H, OUYANG T, et al. In situ exsolved CuZn alloy electrocatalysts for CO2 conversion to tunable syngas production[J]. Advanced Functional Materials, 2025, 35(7): 2415367.
[13] ZHANG K, WANG J, ZHANG W, et al. Adjusted preferential adsorption of intermediates via regulation of the electronic structure during the electrocatalytic CO2 reduction process[J]. The Journal of Physical Chemistry Letters, 2023, 15(1): 34-42.
[14] WANG Y, ZOU S, CAI W B. Recent advances on electro-oxidation of ethanol on Pt- and Pd-based catalysts: from reaction mechanisms to catalytic materials[J]. Catalysts, 2015, 5(3): 1507-1534.
[15] LAI S C S, KLEIJN S E F, ÖZTÜRK F T Z, et al. Effects of electrolyte pH and composition on the ethanol electro-oxidation reaction[J]. Catalysis Today, 2010, 154(1-2): 92-104.
[16] 屈云腾. 乙醇电氧化催化剂制备及其反应机理研究[D]. 黑龙江: 哈尔滨工业大学, 2017.
[17] GOMES J F, BERGAMASKI K, PINTO M F S, et al. Reaction intermediates of ethanol electro-oxidation on platinum investigated by SFG spectroscopy[J]. Journal of Catalysis, 2013, 302: 67-82.
[18] ZHOU Y, BOWERS B, BAGGER A, et al. Revisiting Active Site Quantification in CO2 Electroreduction: the case for CO displacement[J]. ACS Energy Letters, 2025, 10(9): 4324-4331.
[19] QUAN Y, ZHU J, ZHENG G. Electrocatalytic reactions for converting CO2 to value-added products[J]. Small Science, 2021, 1(10): 2100043.
[20] CHEN Q, LIU K, ZHOU Y, et al. Ordered Ag nanoneedle arrays with enhanced electrocatalytic CO2 reduction via structure-induced inhibition of hydrogen evolution[J]. Nano Letters, 2022, 22(15): 6276-6284.
[21] GAO X, JIANG Y, LIU J, et al. Intermediate-regulated dynamic restructuring at Ag-Cu biphasic interface enables selective CO2 electroreduction to C2+ fuels[J]. Nature Communications, 2024, 15(1): 10331.
[22] MA F, ZHANG P, ZHENG X, et al. Steering the site distance of atomic Cu-Cu pairs by first‐shell halogen coordination boosts CO2 -to-C2 selectivity[J]. Angewandte Chemie International Edition, 2024, 63(46): e202412785.
[23] LI J, ZENG H, DONG X, et al. Selective CO2 electrolysis to CO using isolated antimony alloyed copper[J]. Nature Communications, 2023, 14(1): 340.
[24] JIA Y, HSU H S, HUANG W C, et al. Probing the roles of indium oxides on copper catalysts for enhanced selectivity during CO2 -to-CO electrochemical reduction[J]. Nano Letters, 2023, 23(6): 2262-2268.
[25] XUE J, DONG X, LIU C, et al. Turning copper into an efficient and stable CO evolution catalyst beyond noble metals[J]. Nature communications, 2024, 15(1): 5998.
[26] ZHU K, JIA B, CHEN Z, et al. Sn Catalysts with Build-in [NCN]2 - as proton relay for industrial-grade CO2 reduction at low overpotential[J]. Angewandte Chemie, 2025, 137(31): e202507422.
[27] CHEN Z, XIAO Y, QIAO X, et al. Monitoring chalcogenide ions-guided in situ transform active sites of tailored bismuth electrocatalysts for CO2 reduction to formate[J]. Proceedings of the National Academy of Sciences, 2025, 122(10): e2420922122.
[28] ZHAO H, XIE Y, LV B, et al. Achieving current density of 815mA/cm2 for electrochemical CO2 reduction to formate by enhancing *OCHO intermediate adsorption through intercalated Bi single atoms in BiOBr[J]. Applied Catalysis B: Environment and Energy, 2025, 371: 125234.
[29] CAI Y, YANG R, FU J, et al. Self-pressurizing nanoscale capsule catalysts for CO2 electroreduction to acetate or propanol[J]. Nature Synthesis, 2024, 3(7): 891-902.
[30] WU Y, CHEN C, YAN X, et al. Enhancing CO2 electroreduction to CH4 over Cu nanoparticles supported on N-doped carbon[J]. Chemical Science, 2022, 13(28): 8388-8394.
[31] SANG J L, ZHANG S D, LIU Q, et al. Liquid‐liquid interface‐driven reconstruction of CuAg nanocomposites for selective CO2 to C2 H4 electroreduction[J]. Small, 2025, 21(46): e08803.
[32] LIU J, LI P, JIA S, et al. Electrocatalytic CO2 hydrogenation to C2+ alcohols catalysed by Pr-Cu oxide heterointerfaces[J]. Nature Synthesis, 2025, 4: 730-743.
[33] CHOI M, BAE S, KIM Y, et al. Selective formaldehyde condensation on phosphorus-rich copper catalyst to produce liquid C3+ chemicals in electrocatalytic CO2 reduction[J]. Nature Catalysis, 2025, 8: 476-486.
[34] CHALA S A, LIU R, OSEGHE E O, et al. Selective electroreduction of CO2 to ethanol via cobalt-copper tandem catalysts[J]. ACS Catalysis, 2024, 14(20): 15553-15564.
[35] KOOLEN C D, PEDERSEN J K, ZIJLSTRA B, et al. Scalable synthesis of Cu-cluster catalysts via spark ablation for the electrochemical conversion of CO2 to acetaldehyde[J]. Nature Synthesis, 2025, 4(3): 336-346.
[36] CAMPBELL C T, SHI S-K, WHITE J. The Langmuir-Hinshelwood reaction between oxygen and CO on Rh[J]. Applications of Surface Science, 1979, 2(3): 382-396.
[37] BAXTER R, HU P. Insight into why the Langmuir-Hinshelwood mechanism is generally preferred[J]. The Journal of Chemical Physics, 2002, 116(11): 4379-4381.
[38] KITA H, SHIMAZU K, KUNIMATSU K. Electrochemical oxidation of CO on Pt in acidic and alkaline solutions: part Ⅰ. voltammetric study on the adsorbed species and effects of aging and Sn(Ⅳ) pretreatment[J]. Journal of Electroanalytical Chemistry and Interfacial Elecrochemistry, 1988, 241(1-2): 163-179.
[39] AUER A, ANDERSEN M, WERNIG E M, et al. Self-activation of copper electrodes during CO electro-oxidation in alkaline electrolyte[J]. Nature Catalysis, 2020, 3(10): 797-803.
[40] LI A, QIU H, WANG Z, et al. Electrocatalytic conversion of methane to ethanol via promoted ·OH generation in aqueous electrolyte[J]. ACS Sustainable Chemistry & Engineering, 2024, 12(25): 9558-9567.
[41] LIU W, LI H C, LI C, et al. Electrochemically promoted activation of light alkanes at ambient conditions[J]. Angewandte Chemie International Edition, 2025, 64(27): e202507417.
[42] LI A Z, WANG X, LI S, et al. Direct Electrooxidation of ethylene to glycol over 90% faradaic efficiency enabled by Cl-modification of the Pd surface[J]. Journal of the American Chemical Society, 2025, 147(12): 10493-10503.
[43] QIN H, YE Y, LIN G, et al. Regulating the electrochemical microenvironment of Ni(OH)2 by Cr doping for highly efficient methanol electrooxidation[J]. ACS Catalysis, 2024, 14(21): 16234-16244.
[44] ZHU J, LI Z, LI J, et al. Mixed-solvent solvothermal synthesis of metal-organic framework-supported nickel hydroxide toward a novel lattice oxygen-mediated ethanol oxidation pathway[J]. Applied Catalysis B: Environment and Energy, 2025, 382: 126008.
[45] LUO W, TIAN H, LI Q, et al. Controlled electrocatalytic glycerol upgrading to glyceraldehyde in near neutral media by cobalt oxide lattice activation[J]. Angewandte Chemie International Edition, 2025, 64(34): e202505059.
[46] SONG J, XIAO D, HE J, et al. Revealing the electrochemical-autocatalytic coupling mechanism of Cu-based catalysts for high-potential formaldehyde oxidation[J]. Energy&Environmental Science, 2025, 18(12): 6106-6116.
[47] YAO S, PIAO M, SHI W X, et al. Ag-NiO heterojunction with synergistic adsorption of aldehyde/OH-groups for target electrooxidation[J]. ACS Materials Letters, 2025, 7(8): 2702-2707.
[48] BATISTA E A, IWASITA T. Adsorbed intermediates of formaldehyde oxidation and their role in the reaction mechanism[J]. Langmuir, 2006, 22(18): 7912-7916.
[49] LI J, LI L, MA X, et al. Selective ethylene glycol oxidation to formate on nickel selenide with simultaneous evolution of hydrogen[J]. Advanced Science, 2023, 10(15): 2300841.
[50] MA X Y, DING C, LI H, et al. Revisiting the acetaldehyde oxidation reaction on a Pt electrode by high-sensitivity and wide-frequency infrared spectroscopy[J]. The Journal of Physical Chemistry Letters, 2020, 11(20): 8727-8734.
[51] XIAO Y, GUO Z, CAO J, et al. Revealing operando surface defect-dependent electrocatalytic performance of Pt at the subparticle level[J]. Proceedings of the National Academy of Sciences, 2024, 121(22): e2317205121.
[52] TIMOSHENKO J, ROLDAN CUENYA B. In situ/operando electrocatalyst characterization by X-ray absorption spectroscopy[J]. Chemical reviews, 2020,121(2):882-961.
[53] LI F Z, QIN H G, GU J. Development of electrolysis systems for ambient temperature CO2 reduction[J]. EnergyChem, 2025, 7(3): 100156.
[54] YUAN S, WANG R, XUE R, et al. Flow field design matters for high current density zero-gap CO2 electrolyzers[J]. ACS Energy Letters, 2024, 9(12): 5945-5954.
[55] LIU T, WANG Y, WU Y, et al. Continuous decoupled redox electrochemical CO2 capture[J]. Nature Communications, 2024, 15(1): 10920.
[56] LI C, GUO M, YANG B, et al. Efficient and scalable upcycling of oceanic carbon sources into bioplastic monomers[J]. Nature Catalysis, 2025, 8: 1023-1037.


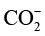 );随后,活化中间体与电解质中的质子(H+)结合,经历多步电子/质子转移(Proton-Coupled Electron Transfer,PCET),最终脱附生成目标产物(如CO、CH4等)。这一过程中,催化剂对中间体的吸附强度至关重要:若吸附过强,会导致中间体难以进一步转化(如CO在Pt表面强吸附易造成催化剂中毒);若吸附过弱,则无法有效活化反应物(如CO2在惰性金属表面难以生成稳定中间体),因此“适当的吸附强度”是实现高效转化的核心条件[7]。
);随后,活化中间体与电解质中的质子(H+)结合,经历多步电子/质子转移(Proton-Coupled Electron Transfer,PCET),最终脱附生成目标产物(如CO、CH4等)。这一过程中,催化剂对中间体的吸附强度至关重要:若吸附过强,会导致中间体难以进一步转化(如CO在Pt表面强吸附易造成催化剂中毒);若吸附过弱,则无法有效活化反应物(如CO2在惰性金属表面难以生成稳定中间体),因此“适当的吸附强度”是实现高效转化的核心条件[7]。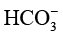 的迁移)也会影响界面电荷传递效率,若电子传导速率与离子迁移速率不匹配,会形成电荷积累或浓度极化,进一步增加反应过电位,降低能量利用效率。
的迁移)也会影响界面电荷传递效率,若电子传导速率与离子迁移速率不匹配,会形成电荷积累或浓度极化,进一步增加反应过电位,降低能量利用效率。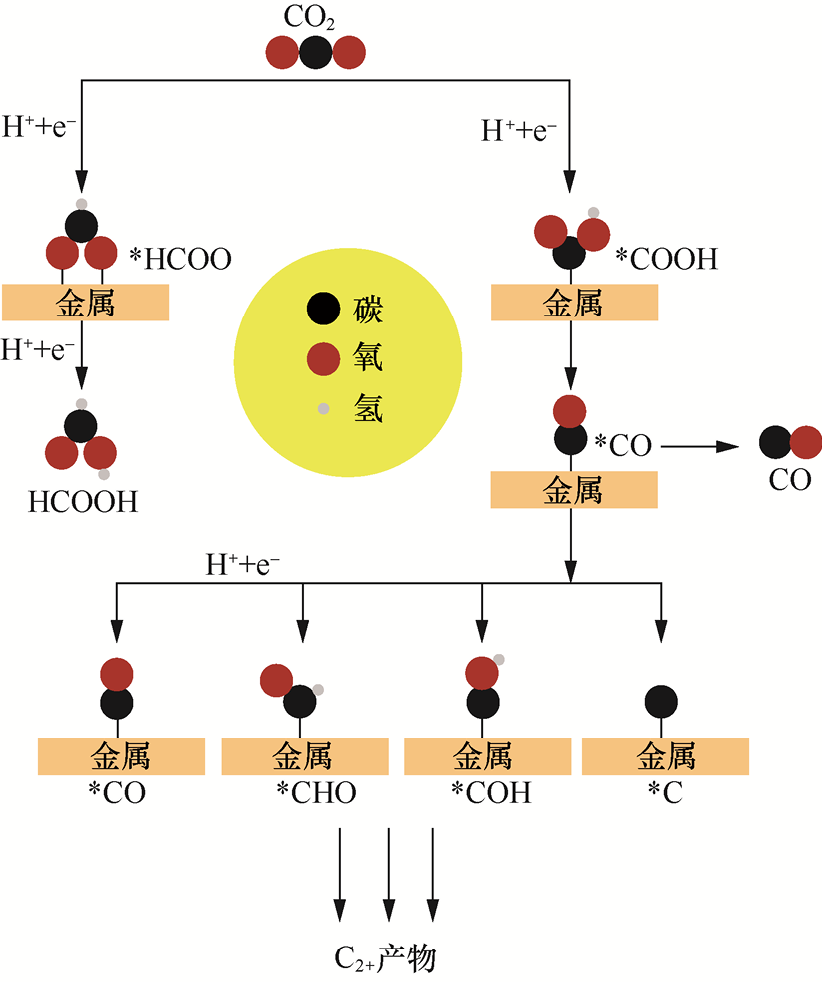
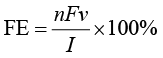
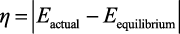
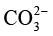 /
/
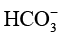 、
、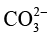 ),而阳极氧析出反应又会造成电解液酸度提高,两侧pH值不平衡将导致催化剂活性衰减。同时,由于装置对电解液量需求较大(单侧通常需50~200mL),产物会被显著稀释,后续分离能耗显著增加。
),而阳极氧析出反应又会造成电解液酸度提高,两侧pH值不平衡将导致催化剂活性衰减。同时,由于装置对电解液量需求较大(单侧通常需50~200mL),产物会被显著稀释,后续分离能耗显著增加。